
Abstract
Rift Valley fever virus (RVFV) causes Rift Valley fever (RVF), resulting in morbidity and mortality in humans and ruminants. Evidence of transboundary outbreaks means that RVFV remains a threat to human health and livestock industries in countries that are free from the disease. To enhance surveillance capability, methods for detection of RVFV are required. The generation of reagents suitable for the detection of RVFV antigen in formalin-fixed, paraffin-embedded tissues from infected animals have been developed and are described herein. Recombinant nucleoprotein (rNP) was expressed in Escherichia coli and purified using immobilized metal ion affinity chromatography. Purified rNP was used as an immunogen to produce anti-NP polyclonal antisera in rabbits for use in detection of RVFV NP in experimentally infected animals by immunohistochemistry. Antisera raised in rabbits against rNP were able to recognize viral NP antigen in fixed infected Vero cell pellets and sheep liver. Therefore, the methods and reagents described herein are useful in assays for detection of RVFV infections in animals, for research and surveillance purposes.
Rift Valley fever virus (RVFV; Bunyavirales, Phenuiviridae, Rift Valley fever phlebovirus) is a zoonotic arthropod-borne phlebovirus 9 that causes Rift Valley fever, a severe disease for which no approved vaccine currently exists for human use. 3 In the United States, RFVF is listed as a Select Agent and Toxin, and the National Institute of Allergy and Infectious Disease lists RFVF as a Category A priority pathogen. 4 In Australia, RVFV is categorized as a Security Sensitive Biological Agent by the federal government. In humans, clinical signs of RVF range from a mild form of febrile illness to a severe form characterized by retinopathy, meningoencephalitis, or hemorrhagic fever; however, the disease is primarily one of sheep, goats, and cattle. 11 Affected animals demonstrate high rates of morbidity and mortality, with younger animals, neonates, and fetuses being most susceptible. 11 Typical signs of RVF include acute fever, anorexia, abortion, or neonatal mortality, but many of the clinical signs of RVF are nonspecific, confounding the ability of veterinarians to make differential diagnoses. 1 Hence, laboratory-based assays, informed by clinical findings, are required to differentiate RVF from diseases with overlapping clinical presentations.
RVFV was confined to Africa until outbreaks in livestock in and around Madagascar and on the Arabian Peninsula were detected, demonstrating the transboundary threat that the disease poses.11,19 The threat of outbreaks in countries that are free of the virus remains high as a result of the movement of livestock, human migration, 8 and the presence of competent vectors such as Aedes, Culex, and Mansonia spp. of mosquitoes in these countries. 17 This persistent threat demands a suitable preparedness response aimed at limiting the impact of the disease if an incursion were to occur.
RVFV infections have been detected by identification of virus or viral antigen, antibody, or RNA. The nucleoprotein (NP) of Bunyavirales is highly conserved among the genus Phlebovirus and is an immunodominant antigen that has produced high antibody titers in multiple species,7,16,18 making it a useful target for NP-specific antibodies. 10 In fact, monoclonal 12 and polyclonal 2 antibodies have been used to detect RVFV infection by detecting NP in fixed tissues. Another favorable aspect of RVFV NP as a target for assay development is its abundant expression during infection, which maximizes the potential sensitivity of the assay. 6 Furthermore, experimentally generated mouse antibodies to non-RVFV Bunyavirales NPs demonstrated negligible cross-reactivity with RVFV NP, suggesting that high assay specificity for RVFV may also be possible using immunoassays. 14 For these reasons, we selected RVFV NP as the target of our work to detect RVFV infections for research and pre- or post-outbreak surveillance purposes.
A representative RVFV NP sequence (AAF00695.1) was used to produce a codon-optimized gene (Atum) inserted into pET-3a that was used to transform Escherichia coli strain BL21 cells (DE3; Thermo Fisher Scientific). An individual transformed colony was grown in lysogeny broth containing 100 µg/mL ampicillin (Merck) and induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG; Promega) for 3 h before cells were harvested at 3,000 × g for 5 min. Inclusion bodies were isolated in the presence of an EDTA-free, protease-inhibitor cocktail (Merck) using BugBuster master mix (BBMM; Merck) then solubilized in binding buffer (50 mM Na2HPO4 pH 8, 300 mM NaCl, 6 M urea, 10 mM 2-mercaptoethanol, 10 mM imidazole, EDTA-free, protease-inhibitor cocktail). Recombinant NP (rNP) was purified by immobilized metal affinity chromatography (IMAC) and quantified (EZQ protein quantitation kit; Thermo Fisher Scientific).
Expressed and purified RVFV rNP (Fig. 1) were boiled for 10 min in lithium dodecyl sulfate (Thermo Fisher Scientific), 50 mM dithiothreitol sample buffer, then resolved electrophoretically at 200 V for 40 min on 4–12% bis-Tris polyacrylamide gels in morpholineethanesulfonic acid buffer (Thermo Fisher Scientific). Coomassie staining of the gels showed that rNP could be purified from the urea-solubilized fraction of the cell lysate by IMAC (Fig. 1). Approximately half of the total expressed rNP was soluble (Fig. 1, lanes 4 and 5) and half of this soluble fraction bound IMAC resin (Fig. 1, lanes 6 and 7, shows unbound fraction). The high purity of the rNP was evident, with no additional protein bands visible by Coomassie brilliant blue staining (Fig. 1, lanes 10 and 11). Overall, without optimization, the system yielded ~ 35 mg of rNP per L of culture, which is comparable to that observed in at least one other study (> 38 mg/L). 5
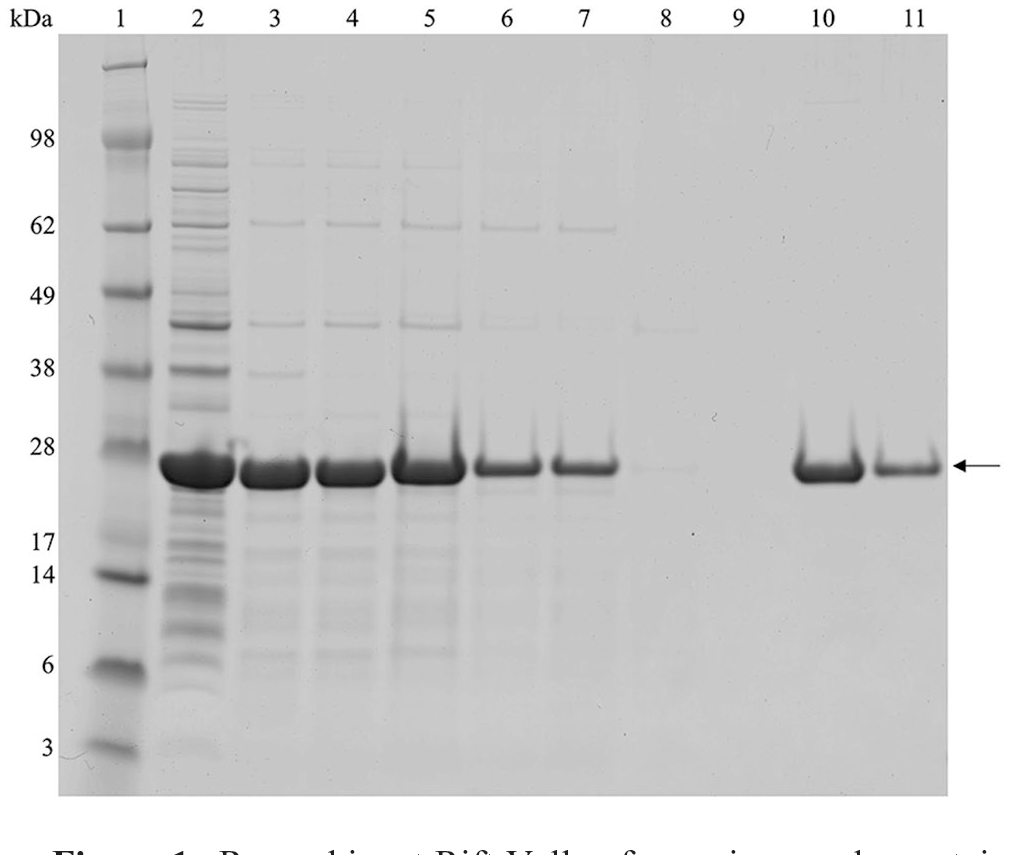
Recombinant Rift Valley fever virus nucleoprotein (NP) purification. Recombinant NP (rNP) purified using immobilized metal affinity chromatography (IMAC) was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and stained with Coomassie brilliant blue. Lane 1 = SeeBlue Plus2 molecular mass marker (Thermo Fisher Scientific); lane 2 = total expressed rNP post-induction culture lysed in BugBuster master mix (BBMM; Merck); lane 3 = BBMM- and urea-insoluble fraction of rNP culture; lane 4 = BBMM-insoluble and urea-soluble fraction of rNP culture; lane 5 = fraction soluble in urea loaded onto the IMAC column; lanes 6, 7 = fractions from 2 applications that were not bound by the IMAC column; lanes 8, 9 = successive column washes; lanes 10, 11 = first and second elutions of purified rNP. Molecular masses are given in kilodaltons (kDa). Arrow indicates rNP.
Purified rNP was then used to generate polyclonal antisera, with approval from the Australian Animal Health Laboratory (AAHL) Animal Ethics Committee and the Department of Agriculture and Water Resources (In Vivo Approval). Two New Zealand White rabbits were immunized twice with 75 µg rNP in adjuvant, 15 18 d apart. Two weeks later, both rabbits were exsanguinated under anesthetic to yield a total of ~ 125 mL of hyperimmune antiserum. The decision to produce and use polyclonal antisera for immunohistochemical applications rather than monoclonal antibodies was based upon the rapidity and ease of production, higher avidity, and general robustness for immunostaining of formalin-fixed tissues. To determine if the sera were able to recognize rNP and RVFV NP from infected cells, these proteins were transferred to a polyvinylidene fluoride membrane in CAPS (3-(cyclohexylamino)-1-propanesulfonic acid)–methanol buffer (pH 11; Bio-Rad Laboratories). Membranes blocked with 5% (w/v) skim milk were probed with rabbit anti–RVFV rNP antisera diluted 1:10,000 followed by goat anti-rabbit IgG–horseradish peroxidase conjugate (Bio-Rad Laboratories) diluted 1:20,000. Sera reacted visibly with rNP down to 10 ng (Fig. 2). The antisera also reacted strongly with NP from 4 different South African RVFV isolates present in tissue culture supernatant from infected Vero (African green monkey kidney) cells (Fig. 2).
Immunoreactivity of rabbit anti–Rift Valley fever virus (RVFV) nucleoprotein (NP) polyclonal antisera with recombinant and endogenous RVFV NP. Lane 1 = Precision Plus molecular mass marker (Bio-Rad Laboratories); lanes 2–6 = 100, 75, 50, 25, and 10 ng rNP, respectively; lanes 7–10 = tissue culture supernatant containing South African RVFV isolates SA 152/08, SPU 86/09, SA 276/10, and SA 633/10, respectively. Molecular masses are given in kilodaltons (kDa). Arrow indicates rNP or RVFV NP.
Next, we investigated the ability of the anti-rNP antibodies to recognize RVFV NP in formalin-fixed, paraffin-embedded (FFPE) infected cell pellets. Uninfected Vero cells or those infected with RVFV South African strain SPU 86/09 for 48 h were fixed with 10% neutral-buffered formalin for at least 30 min then pelleted by centrifugation and resuspended in molten neutral agar. Once cooled, the samples were embedded with paraffin wax using a bench-top tissue processor (TP1020; Leica Microsystems) then sectioned (4 µm) and dewaxed in preparation for immunohistochemistry (IHC). A titration of rabbit sera used to immunostain Vero cells infected with RVFV showed strong specific intracytoplasmic staining at a serum dilution of 1:1,000 (Fig. 3). The intensity of staining was unchanged for dilutions ranging from 1:400 to 1:1,600; however, some background staining was visible at dilutions lower than 1:800 (data not shown). Uninfected cells showed no staining when sera diluted 1:1,000 were used (Fig. 3). Successful immunostaining of formalin-fixed cultured cells suggested the antisera would be useful for immunocytochemical detection of RVFV antigen.
Immunocytochemistry and immunohistochemistry using rabbit anti–Rift Valley fever virus (RVFV) nucleoprotein (NP). Vero cells (upper images) or sheep liver sections (lower images) were tested for RVFV antigen. No antigen was present in uninfected cells or liver sections from an uninfected sheep (left images); sections from infected cells or liver (right images) showed dense intracytoplasmic antigen staining in hepatocytes. Vero cell pellets and liver sections were probed with anti–recombinant NP serum from rabbit 2 diluted 1:1,000 followed by anti-rabbit horseradish peroxidase. Lillie–Mayer hematoxylin counterstain.
With approval from the AAHL Animal Ethics Committee, animal studies were conducted in accordance with the Australian code for the care and use of animals for scientific purposes. Four 2-mo-old Merino sheep were experimentally inoculated with RVFV strain SPU 86/09, originally isolated from a South African bovid in 2009. A total dose of 106 TCID50 was injected subcutaneously. Two sheep (sheep 1 and 3) that developed acute disease with fever and lethargy were euthanized 3 d after challenge. Two other sheep (sheep 2 and 4) developed fever and mild disease and thereafter recovered; sheep 2 had transient fever at d 2 after challenge; sheep 4 had intermittent fever between d 1 and 9 post-challenge; these sheep were euthanized while clinically healthy 17 d after challenge.
IHC is an important tool for research and detection of RVFV infection. RVFV affects multiple organs of infected ruminants including liver, lymph nodes, kidney, spleen, and lung. 13 Infection of the liver is well characterized and has been used extensively to facilitate diagnoses of RVF. For these reasons, liver samples from the experimentally infected sheep were selected as representative tissues for testing the efficacy of the rabbit anti-rNP sera in the detection of viral NP in FFPE tissue. Liver sections from sheep 1 acutely infected with RVFV and from a naïve sheep were prepared for IHC as above, except antigen retrieval at 97°C and pH 9 was performed for 20 min (PT link system; Agilent) before quenching with 3% (w/v) H2O2 for 10 min. Sections were incubated under 200 µL of rabbit anti-rNP serum diluted 1:1,000 for 45 min followed by 200 µL of EnVision HRP-conjugated anti-rabbit IgG (Agilent) used neat for 20 min. Chromogen development was performed with 3-amino-9-ethylcarbazole (AEC) substrate-chromogen (Agilent) for 10 min, and sections were counterstained with Lillie–Mayer hematoxylin for 30 s followed by brief submersion in Scott tap water. Stained slides were mounted (Faramount aqueous mounting medium; Agilent) and examined with a light microscope (DM2000; Leica).
Strong immunostaining of intracytoplasmic antigen was identified in hepatocytes within the periacinar areas of the infected liver taken from sheep 1 during acute infection (Fig. 3). Immunostaining was associated with severe acute periacinar hepatocellular necrosis, a lesion that is typical of that described for RVF in sheep (Fig. 3). Liver sections from uninfected sheep showed no staining for NP using antisera diluted 1:1,000 (Fig. 3). Thus, typical RVFV staining was observed in liver from the infected sheep, suggesting that the antisera would be useful for IHC detection of RVFV in sheep experimentally or naturally infected with RVFV.
We set out to enhance preparedness for a RVFV incursion by developing methods that, in the first instance, would lead to the development and production of reagents suitable for the immunohistochemical detection of RVFV infection in experimentally infected cells and animals. To this end, described herein are methods for the production of rNP protein and polyclonal sera raised to specifically recognize RVFV NP. Our processes yielded 35 mg rNP/L of culture, providing sufficient antigen to raise at least 14.5 L of antiserum for the performance of at least 72 million immunohistochemical detection tests. This pilot procedure alone has generated sufficient antisera for 625,000 individual IHC tests, enabling our laboratory to respond effectively to any future incursion of RVFV that may occur in Australia.
Footnotes
Acknowledgements
We thank David Williams and Jeff Butler (AAHL) for critical reading of the manuscript; Drs. Janusz Paweska and Petrus Jansen van Vuren (National Institute for Communicable Diseases, Johannesburg, South Africa) for the provision of strains of RVFV; Animal Services Teams at AAHL for assistance with the animal studies; and the Histology team at AAHL for histology support.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding was provided by the Australian Government, Department of Agriculture, which had no further involvement in the study.