
Abstract
We characterized the antibody response to decorin-binding protein A (DbpA) or DbpB from immune serum samples collected from 27 dogs infected with Borrelia burgdorferi by Ixodes scapularis ticks. Immunoglobulin M (IgM) antibodies to DbpA or DbpB were rarely detected, but high levels of IgG antibodies to DbpA were detected in 16 of 27 of the immune sera collected 1 mo after infection, 20 of 25 of the sera collected after 2 mo, and each of the 23, 17, or 11 serum samples evaluated after 3, 4, or 5 mo, respectively. In addition, IgG antibodies to DbpB were detected in 22 of 27 (p = 0.005) tested dogs after 1 mo, and the frequency of detecting the antibodies thereafter closely mimicked the antibody responses to DbpA. Moreover, antibodies to DbpA or DbpB were not produced by dogs vaccinated with a whole-cell B. burgdorferi bacterin; removing the antibodies to DbpA by adsorption to recombinant DbpA (rDbpA) did not affect the reactivity detected by a rDbpB ELISA. Therefore, the findings from our preliminary study showed that antigenically distinct antibodies to DbpA or DbpB are produced reliably during canine infection with B. burgdorferi, and the response is not confounded by vaccination with a Lyme disease bacterin. Larger studies are warranted to more critically evaluate whether detecting the antibody responses can improve serodiagnostic confirmation of canine Lyme disease.
Lyme disease, caused primarily in the United States by the transmission of Borrelia burgdorferi from infected Ixodes scapularis ticks, can cause morbidity in dogs. Although infected dogs more typically suffer from chronic subclinical polyarthritis, 19 untreated infections may also cause frank arthritis, 12 renal failure, 8 or cardiac arrhythmias. 11 Infection with B. burgdorferi can be confirmed by culture or PCR. However, recovering the organisms by culture is confounded by low sensitivity and the need for specialized growth medium and long incubation periods. 20 In addition, detecting B. burgdorferi DNA by PCR is hindered by the low number of spirochetes in clinical specimens. 7 Therefore, providing indirect evidence of infection by detecting antibodies formed against the spirochetes is, to date, the most common laboratory method for detecting canine Lyme disease.
Serodiagnostic tests for confirming canine infection with B. burgdorferi rely most often on detecting antibodies to a synthetic peptide (C6) derived from the Borrelia spp. variable surface–expressed (VlsE) antigen, outer surface protein C (OspC), and/or OspF.5,13,15,21 Although each can provide confirmation of infection, several shortcomings remain problematic. For example, persistence of the antibodies to C6 for months to years after successful therapy 16 confounds the ability to discriminate active infection from past exposure. The OspC used for laboratory testing may not contain the epitope targeted by the antibodies to OspC, given that the ospC gene is extremely heterogeneous, even among isolates from the same geographic region, 1 and commercial Lyme disease bacterins also induce antibodies to OspC.10,15 In addition, antibodies to OspF are not always produced by B. burgdorferi–infected dogs. 5 Therefore, identifying additional antibody responses that are reliably produced during canine infection with B. burgdorferi, especially responses that are not confounded by Lyme disease vaccines, are important because their identification may then induce further investigation to determine whether the response can be used to improve the diagnostic regimen.
Researchers have shown that immunoglobulin G (IgG) antibodies to an ~ 20-kDa B. burgdorferi protein were universally produced by laboratory dogs infected with Lyme disease spirochetes and not by recipients of a Lyme disease bacterin. 10 We sought to determine the specificity of the antibodies by using archived serum samples from previously published5,10 studies that had been approved by the Animal Care and Use Committee at Merck Animal Health. Serum samples had been obtained immediately prior to challenging 27 laboratory Beagles by attaching 10 female and 10 male I. scapularis ticks infected with B. burgdorferi to each animal and allowing the female ticks to feed to repletion; additional serum samples were collected monthly for 5 mo. In addition, infection with B. burgdorferi had been confirmed in each dog by recovering the spirochetes from the skin biopsies of 14 dogs and the skin biopsies and joint tissues from 13 dogs. Since completing the earlier studies, however, only various amounts of the immune sera, including 27 serum samples collected pre-tick challenge or 1-mo post-tick challenge and 25, 23, 17, and 11 samples collected 2, 3, 4, or 5 mo post-tick challenge, respectively, had been stored at −20°C without thawing. The numbers of sera were decreased at each successive time because dogs had been removed from the previous studies5,10 for autopsy and culture when the animal developed Lyme disease–related clinical signs. Therefore, we used the remaining archived serum samples to more critically evaluate the antibodies that bound the 20-kDa B. burgdorferi protein.
We initially determined whether the IgG antibodies to the 20-kDa protein described previously 10 could be detected in a subset (n = 20) of the 27 immune serum samples collected 3 mo post-tick challenge by western blotting. Sampling only a subset was necessary because the volumes of the remaining 7 samples from that time were extremely limited and we wished to preserve adequate amounts for additional evaluation. Briefly, B. burgdorferi sensu stricto 297 spirochetes were cultured at 34°C in Barbour–Stoenner–Kelly (BSK) medium until reaching logarithmic growth, and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blot was performed using standard techniques. The washed and concentrated logarithmic phase spirochetes were resuspended in 2× sample buffer (65.8 mM Tris-HCl [pH 6.8], 26% glycerol, 100 mM dithiothreitol, 2.1% sodium dodecyl sulfate, 0.001% bromophenol blue), boiled for 5 min, and then loaded onto a 10–20% gradient polyacrylamide gel (Bio-Rad, Hercules, CA). The gel was then electrophoresed, and the proteins were transferred onto a 0.45-µM polyvinylidene difluoride membrane by electrophoresis. The membranes were subsequently cut into strips and incubated with immune serum diluted 1:100 with pH 7.2 phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBST) followed by goat anti-dog IgG antibodies conjugated with horseradish–peroxidase; the bound antibodies were detected by using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate.
When the immune sera were diluted 100-fold with PBST, the evaluation showed that IgG antibody bands corresponding to multiple B. burgdorferi proteins were detected, and the response included without exception an intense band that corresponded to the ~ 20-kDa protein. Because the 20-kDa protein reactive band was so intense, we re-evaluated several of the immune sera by repeating the western blot evaluation using more dilute sera (1:1,000 with PBST), and the antibody levels were then decreased enough to show that the original “intense” band was more accurately comprised of 2 bands that corresponded to 19-kDa and 20-kDa proteins.
The finding that the antibodies were binding both a 19-kDa and 20-kDa B. burgdorferi protein provided significant evidence that the response was targeting decorin-binding proteins A (DbpA) and DbpB. In support, the molecular weights of each are similar, the proteins are co-transcribed by a 2-gene operon encoded on a 54-kb (lp54) linear plasmid,9,22 each is expressed on the surface of viable spirochetes during mammalian infection,6,18 and both perform a critical role during colonization of the mammalian host. 17 In addition, numerous researchers2,6,18,22 had confirmed that antibodies to DbpA and DbpB antibodies were reliably produced during human Lyme disease and that DbpB was an important antigen during canine B. burgdorferi infection. 3 We, therefore, confirmed this possibility by re-evaluating the 20 immune sera by western blot, but we used B. burgdorferi sensu stricto BbKH500 as the antigen, which lacks expression of DbpA or DbpB as a result of the insertion of kanr into the dpbBA operon. 4 In each instance, the numerous band reactivities detected by using the 297 spirochetes remained detectable but, without fail, the antibodies that bound 19-kDa or 20-kDa proteins were absent.
Spurred by confirmation that the antibodies were specific for DbpA and DbpB, we next prepared recombinant DbpA (rDbpA) and rDbpB ELISAs and evaluated the reactivities in the pre-tick challenge sera and the sera collected from the various times after infection with the Lyme disease spirochetes. To accomplish this, DNA was recovered from a fresh culture of B. burgdorferi sensu stricto B31 (UltraClean microbial DNA isolation kit; Qiagen, Germantown, MD), and the following were used to amplify each gene: forward primer 5′-CACCGGACTAACAGGAGCAACAAAAA-3′and reverse primer 5′-TTAGTTATTTTTGCATTTTTCATC-3′ for dbpA; forward primer 5′-CACCAGTATTGGATTAGTAGAAAGAA-3′ and reverse primer 5′-TTATTTCTTTT-TTTTGCTTTTATT-3′for dbpB; and cycling parameters of 98°C for 30 s, 35 cycles at 98°C for 10 s, 60°C for 15 s, 72°C for 15 s, and a final extension cycle at 72°C for 5 min.
Each amplified gene product was then ligated into plasmid pET100/D-TOPO (Thermo Fisher Scientific, Waltham, MA) and transformed into Escherichia coli TOP10 (Thermo Fisher). The transformed cells were subsequently incubated for 2 h at 37°C in 10 mL of 2× YT broth with 1% glucose and 50 µg/mL of ampicillin; a 1-mL volume of each culture was transferred to 500 mL of fresh 2× YT medium with glucose and ampicillin and incubated at 37°C for an additional 2 h. Five hundred microliters of 1 M isopropyl β-D-1-thiogalactopyranoside (IPTG) was added, and the culture was re-incubated at 26°C for 24 h. The recombinant proteins were then recovered by using a commercial kit according to the manufacturer’s recommendations (ProBond purification system; Thermo Fisher Scientific). Briefly, the cell suspension was pelleted by centrifugation, and the cell pellet was lysed by suspension in guanidinium lysis buffer followed by rocking at room temperature for 10 min prior to sonication. The cellular debris was then re-pelleted by centrifugation, and the supernatant was transferred to a column that contained nickel-charged affinity resin to bind the polyhistidine tag on the N-terminus of the recombinant proteins. The bound protein was subsequently washed by passing two 4-mL volumes of pH 6.0 wash buffer followed by six 4-mL volumes of pH 5.3 wash buffer, and then eluted by passing 5 separate 1-mL volumes of elution buffer (pH 4.0) over the column. Each recombinant protein was then dialyzed and concentrated (Amicon centrifugal filter unit; Millipore Sigma, St. Louis, MO); acceptable purity was confirmed by detection of a single band by SDS-PAGE.
Individual wells of microtiter plates were then coated with 100 nanograms of either rDbpA or rDbpB contained in carbonate buffer (90 mM NaHCO3, 60 mM Na2CO3, pH 9.6) prior to incubating the plates overnight at 4°C. Following incubation, the plates were washed with pH 7.2 PBST and then blocked with PBST that contained 1% bovine serum albumin. After rinsing, 100-µL volumes of immune serum diluted 1:200 with PBST were added to each well, and the plates were washed and incubated with peroxidase-conjugated goat anti-dog (SeraCare, Milford, MA) IgM diluted 1:5,000 or IgG diluted 1:25,000 with PBST. The plates were finally washed thoroughly with PBST, o-phenylenediamine substrate (Millipore Sigma) was added, and the absorbances at 490 nm were determined with a spectrophotometer.
To obtain significant cutoff values, we first calculated the mean optical densities (ODs) obtained by testing the pre-tick challenge sera (n = 27), and then considered a value ≥ 3 standard deviations above the mean OD value as significant (> 99%) reactivity. We then compared the findings to those obtained from the post-tick challenge serum samples. As controls, we eliminated interassay variability as a confounding factor by testing the pre- and post-tick challenge samples from each dog on the same ELISA plate and ensured reproducibility by evaluating each serum 3 times using freshly prepared reagents and ELISA plates.
Only minimal amounts of IgM antibodies to DbpA (Fig. 1A) or DbpB (Fig. 2A) were detected in sera collected 1-mo post-tick challenge, and the amount or frequency of detection did not increase significantly at later times. A likely possibility for the lack of reactivity is that the sera were not collected during the peak IgM antibody response. In support, researchers 2 have shown that the peak IgM antibody response to DbpA or DbpB is detected when infected humans have an erythema migrans skin lesion, which typically occurs within the first 2 wk of infection. In contrast, however, high levels of IgG antibodies to DbpA were detected in 16 of the 27 immune sera collected 1 mo after tick challenge, and the antibody response expanded to include 20 of the 25 immune sera evaluated after 2 mo and the sera from each dog infected for ≥ 3 mo (Fig. 1B). In addition, high levels of IgG antibodies to DbpB antibodies were detected in 22 of 27 dogs (p < 0.005, Student t-test) tested 1 mo after infection, and the response also expanded thereafter to include 23 of the 25 dogs evaluated after 2 mo, 22 of the 27 dogs tested after 3 mo, 17 of the 17 dogs tested after 4 mo, and 11 of 11 dogs evaluated after 5 mo (Fig. 2B).
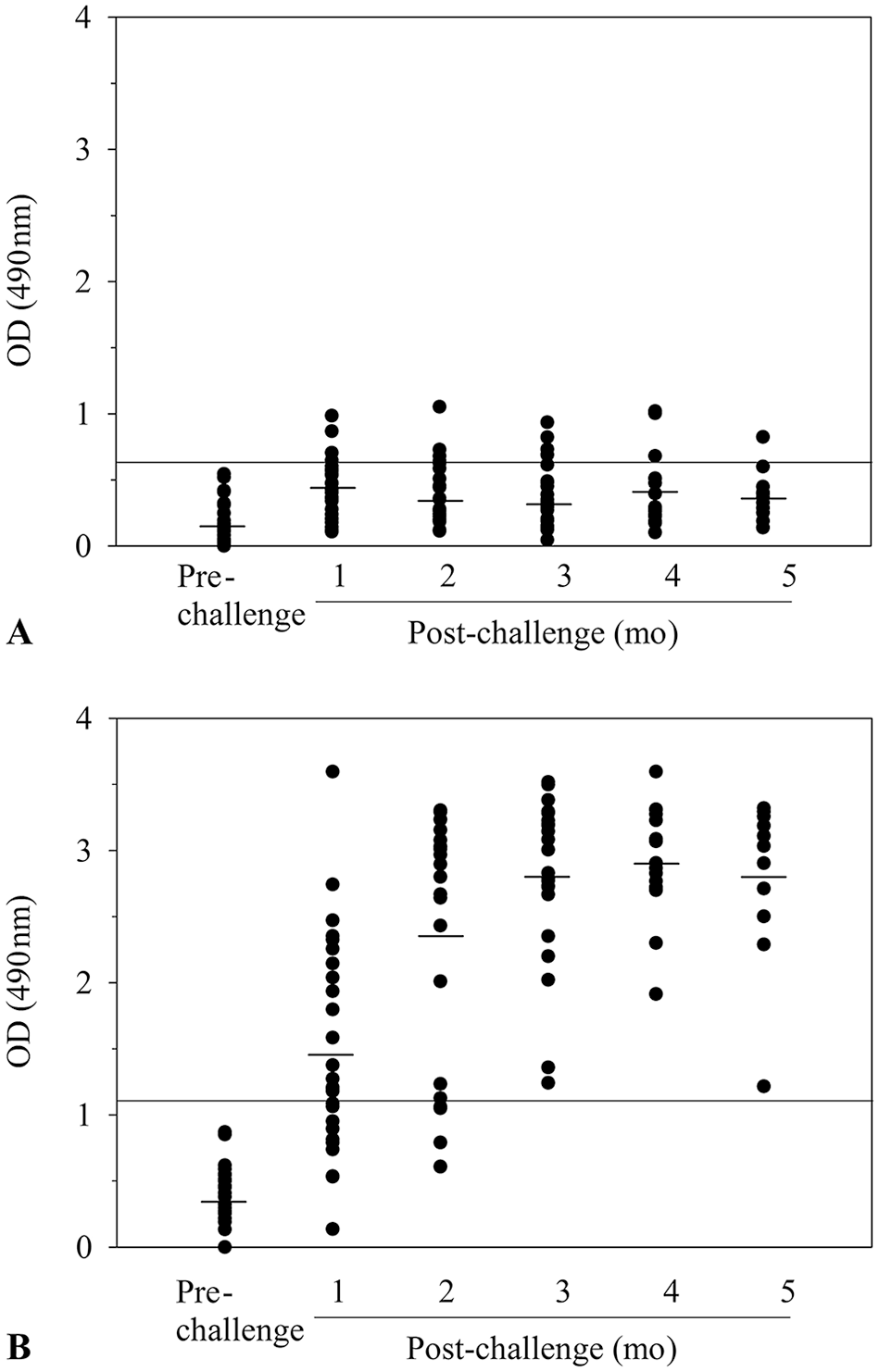
Detection of IgM
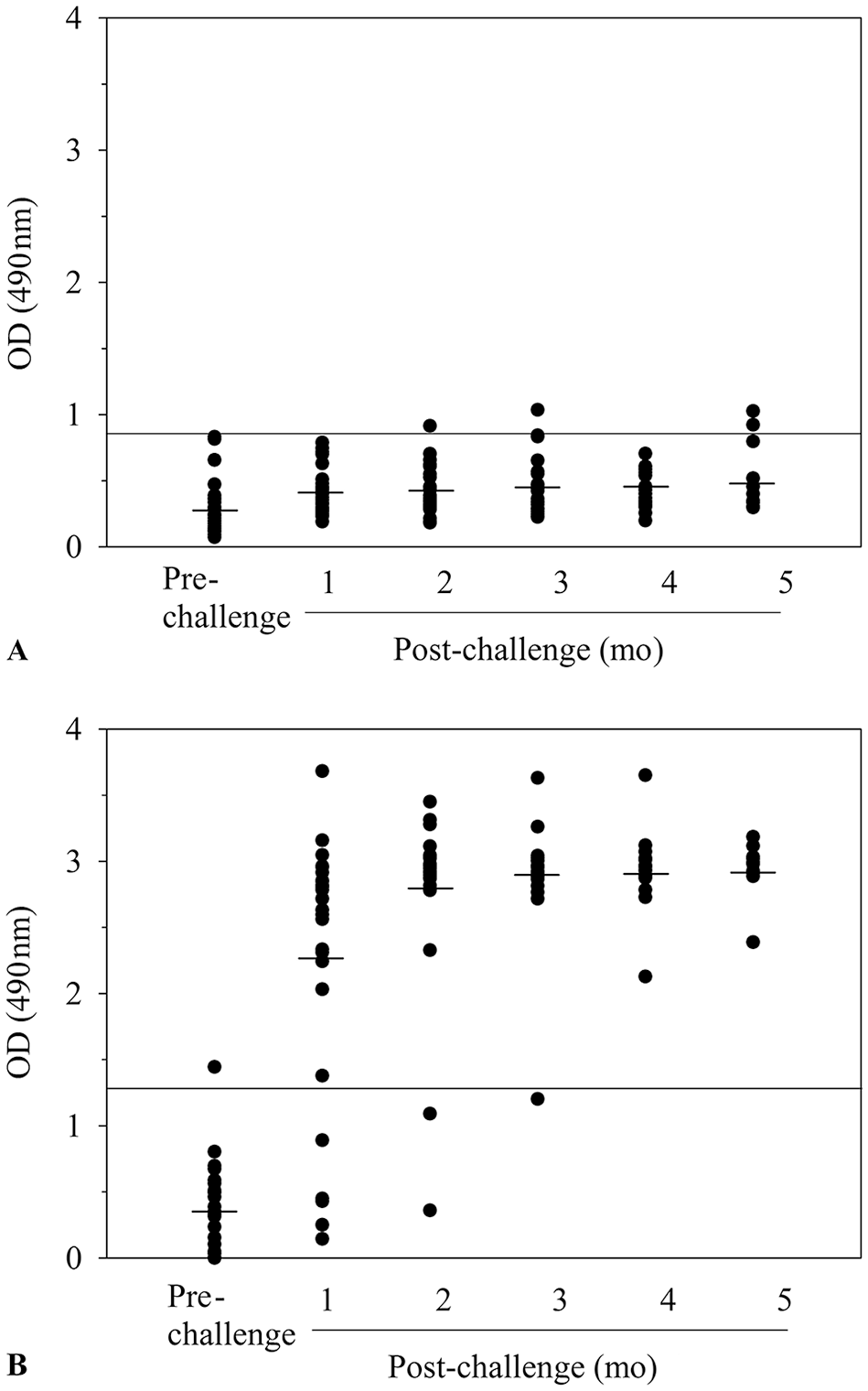
Detection of IgM
Our findings, therefore, showed that the antibody response previously 10 attributed to a 20-kDa B. burgdorferi protein was more accurately antibodies to DbpA and DbpB, and IgG antibodies against each were reliably produced by B. burgdorferi–infected dogs. This is important because, despite studies confirming that antibodies to DbpA or DbpB are important responses during human infection,2,22 the antibody responses during canine infection have not been well-characterized and, as a consequence, neither response is commonly utilized as a diagnostic marker in the United States. 14 Our results, however, support a previous report 3 that DbpB is an important antigen during canine infection and extend this finding by showing that antibodies to DbpA are also produced reliably. Therefore, although we examined the response in only a small number of dogs, the findings should be sufficient to provide the impetus for larger studies that more critically evaluate whether the responses can be used to overcome some of the shortcomings of the current canine Lyme disease diagnostic armamentarium.
Spurred by this possibility, we also performed 2 additional very preliminary experiments to further evaluate the possible diagnostic utility of the antibodies. Because the kinetics of the responses were so similar, we determined whether the antibodies were targeting distinct epitopes by removing the antibodies to DbpA from 5 immune sera by passage over a column that contained 0.5 mg of rDbpA bound to CNBr-activated Sepharose beads (GE Healthcare, Chicago, IL), and then evaluated the effect of removing the antibodies on the reactivities detected by each ELISA. Prior to removing the antibodies to DbpA, the serum samples each contained significant levels of antibodies to DbpA (titers 1:6,400–1:12,800) and DbpB (titers 1:3,200–1:25,600; Table 1). After the DbpA antibodies were removed, the reactivities detected by the rDbpA ELISA were almost completely abrogated (titers < 1:200), whereas the reactivities detected by the rDbpB ELISA remained unaffected.
Effect of removing antibodies to decorin-binding protein A (DbpA) antibodies on ELISA reactivities.
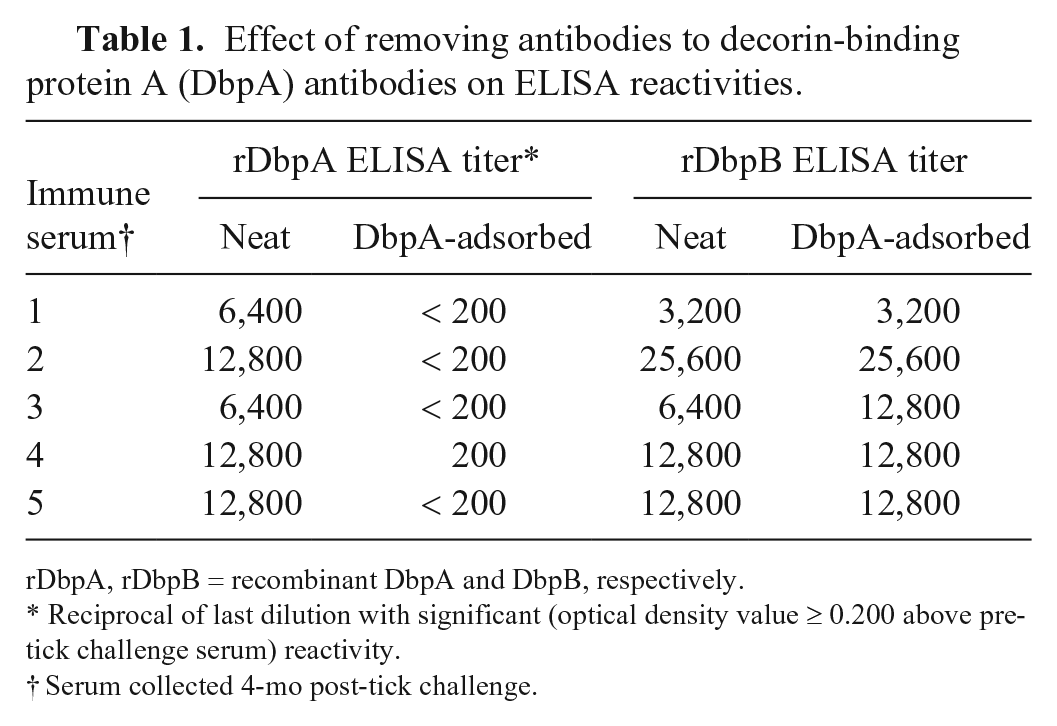
rDbpA, rDbpB = recombinant DbpA and DbpB, respectively.
Reciprocal of last dilution with significant (optical density value ≥ 0.200 above pre-tick challenge serum) reactivity.
Serum collected 4-mo post-tick challenge.
In addition, we performed a cursory evaluation to determine whether previous vaccination with a whole-cell Lyme disease bacterin could be expected to interfere with detection of the antibody responses by testing serum samples collected from 10 dogs vaccinated and boosted one month previously with Nobivac Lyme bacterin (Merck Animal Health, Elkhorn, NE). A previous study 10 had confirmed that each serum sample contained high levels of antibodies to multiple B. burgdorferi proteins but, in each instance, antibodies to DbpA or DbpB were not detected (titers < 1:200) by the ELISAs. Therefore, based on evaluation of only a small number of immune sera from a single time, vaccination with the whole-cell bacterin did not induce potentially confounding antibody responses.
Footnotes
Acknowledgements
We thank Dr. Jon Blevins (University of Arkansas-Fayetteville) for providing B. burgdorferi KH500 and Dr. Andrew Borgert (Gundersen Health System) for statistical analysis.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding was provided by the Gundersen Medical Foundation (La Crosse, WI).