
Abstract
We screened 104 snakes with respiratory disease, collected from 52 snake farms in Guangxi Province, China, for pathogens. Ferlaviruses were detected in 70 of 104 lung samples by reverse-transcription PCR; 34 of 52 of the snake farms were positive for ferlaviruses. No reovirus, adenovirus, sunshine virus, or nidovirus was detected in any of the snakes. We obtained 96 bacterial isolates from snake organs, of which the most commonly isolated species were Salmonella (18) and Proteus (16). Sequence analysis, based on 27 partial RNA-dependent RNA polymerase gene (L) sequences, revealed that ferlaviruses from Guangxi and the known GenBank strains clustered together and formed 3 genogroups. The nucleotide and deduced amino acid homologies of ferlaviruses were 84.3–100% and 95.0–100% within groups, respectively, and 77.0–81.6% and 90.4–95.2% between groups, respectively. Ferlaviruses from Guangxi had close genetic relationships with the known GenBank strains. Our results indicate that ferlaviruses are common in snakes with respiratory disease on the farms of Guangxi that we sampled, and that ferlavirus molecular epidemiology is both diverse and complex.
Respiratory diseases are common in captive snakes, and such diseases usually result in high morbidity and mortality. 18 The genus Ferlavirus (Paramyxoviridae, subfamily Orthoparamyxovirinae) is considered to be an important cause of respiratory disease in snakes. 8 Ferlaviruses are negative-sense, single-stranded RNA viruses with some characteristics of their reptile hosts, including low replication temperatures and a unique unknown (U) gene between the nucleoprotein (N) and phosphoprotein (P) genes. 12 Based on phylogenetic analysis of the L gene, ferlaviruses can be classified into 4 genogroups: groups A–C and a separate cluster consisting of a tortoise isolate.1,2,14
The first recognized ferlavirus outbreak occurred in a Swiss serpentarium in 1976, and the isolate, named Fer de Lance virus (Reptilian ferlavirus), was designated as the type species of the Ferlavirus genus.3,4 Ferlaviruses have been detected, or isolated, in many species of snakes, including Colubridae, Elapidae, Viperidae, Boidae, and Pythonidae. 8 The clinical signs observed are mainly respiratory and occasionally neurologic.
Guangxi Province, located in southern China, has hundreds of private snake farms. Farmed species include the oriental rat snake (Ptyas mucosus, Colubridae), cobra (Naja, Elapidae), and king rat snake (Elaphe carinata, Colubridae). Respiratory diseases are common in the snake farms of Guangxi, and these diseases result in substantial losses. 6 Although ferlavirus is an important pathogen causing respiratory disease in captive snakes in Europe and the United States, little information is available on the epidemiology of this virus in China. 8
We used reverse-transcription PCR (RT-PCR) targeting the L gene to detect ferlaviruses from lung samples of snakes with respiratory disease (Table 1). Partial L gene sequences of ferlaviruses from Guangxi were obtained and compared with known strains from GenBank to evaluate their genetic relationships and epidemic patterns. In view of the possibility of concurrent infection with other pathogens, we also screened for bacteria and other viruses, including reovirus, adenovirus, sunshine virus, and nidovirus. Sunshine virus (Reptile sunshinevirus 1) has only been reported in Australian pythons; nidovirus has been described in pythons from Europe and the United States.7,13,19 In China, no information has been reported on these 2 viruses in snakes.
PCR amplification primer pairs, target genes, and product sizes of ferlavirus, reovirus, adenovirus, sunshine virus, and nidovirus.

In our study, clinical signs in the snakes with respiratory disease mainly included mouth breathing, dyspnea, respiratory noise, and mucus in the mouth; pathologic changes at autopsy included mucoid or caseous exudate in the trachea or lung, pulmonary congestion, pulmonary edema, and pulmonary hemorrhage (Fig. 1). 18 From April 2014 to April 2015, 104 snakes with respiratory disease (74 oriental rat snakes, 26 cobras, and 4 king rat snakes) were screened for pathogens at the Laboratory of Veterinary Pharmacology and Pathology in Guangxi University. Snakes were obtained from 52 snake farms (1–3 snakes per farm) located in Nanning, Chongzuo, Yulin, Qinzhou, Beihai, Baise, and Wuzhou of Guangxi (Table 2). Swabs for bacterial culture were collected from livers, lungs, and hearts, and bacteria were identified following standard protocols. 5 Lungs were collected for PCR analysis.
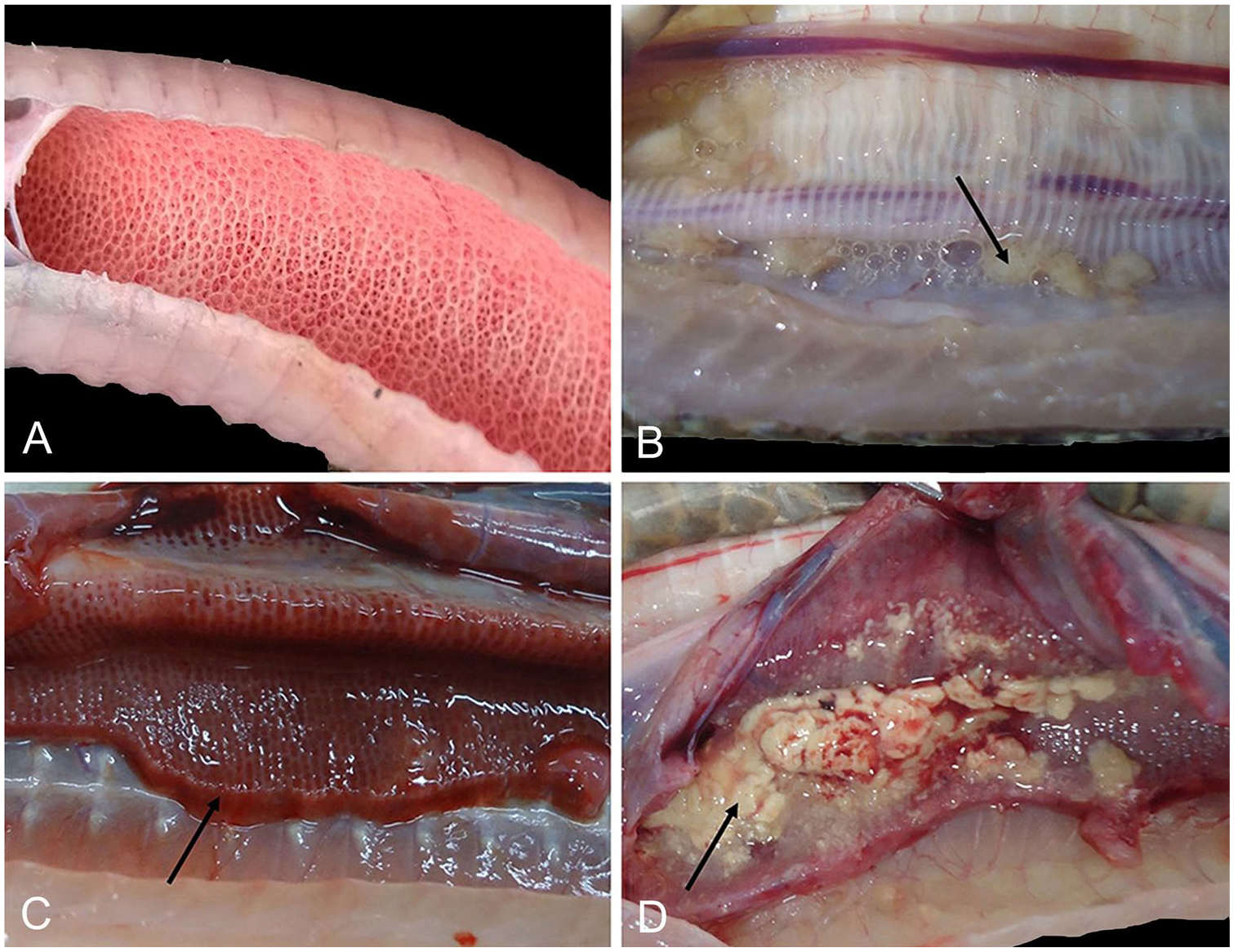
Pathologic findings in snakes with respiratory disease.
Occurrence of ferlaviruses in different regions of Guangxi Province, China.
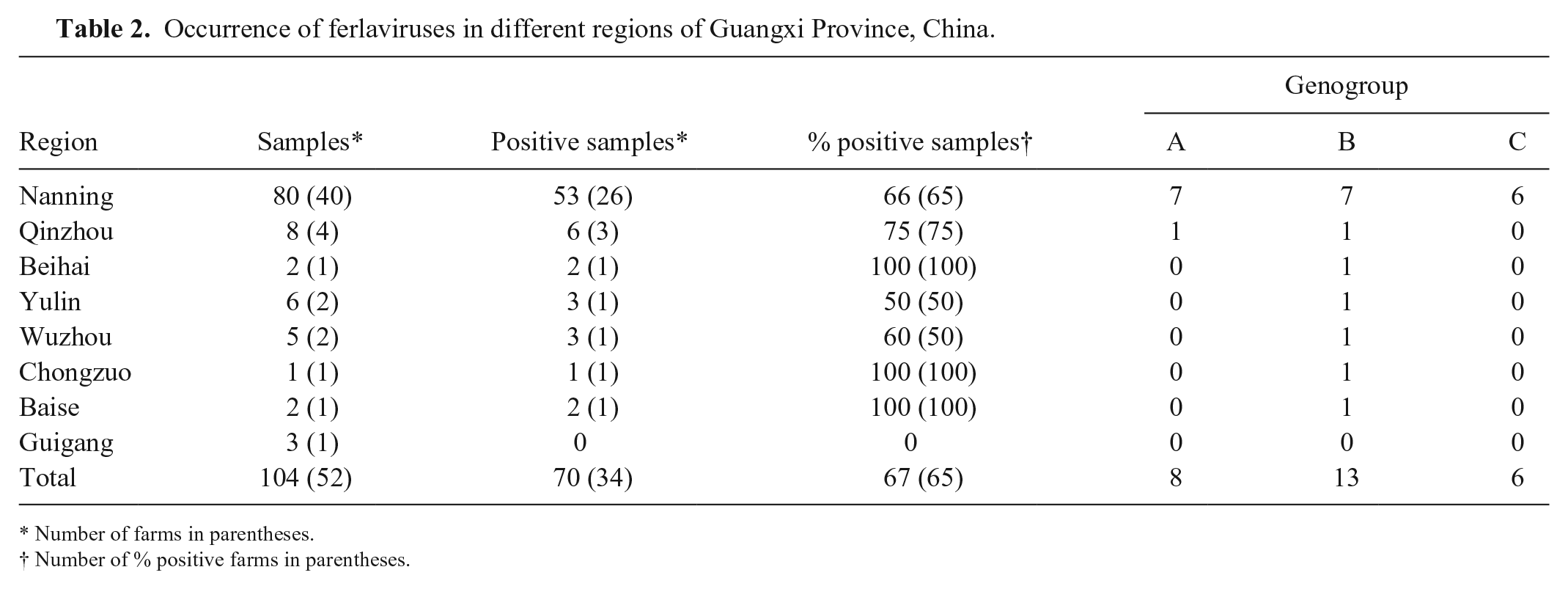
Number of farms in parentheses.
Number of % positive farms in parentheses.
Collected samples were minced with sterilized scissors, placed in 3 mL of phosphate-buffered saline, then frozen and thawed 3 times. Cell debris was pelleted by low-speed centrifugation (1,500 × g for 10 min at 4°C). Total DNA and RNA were extracted from the supernatant (Universal genomic DNA kit; Trizol; CW Bio, Beijing, China), following the manufacturer’s instructions. Extracted RNA was reverse transcribed into complementary DNA (cDNA) using random hexamers and a SuperRT cDNA kit (CW Bio) following the manufacturer’s instructions. Obtained DNA and cDNA were used as templates for PCR amplification.
Primers for the detection of ferlavirus, reovirus, adenovirus, sunshine virus, and nidovirus were synthesized by Beijing Genomics Institute (Beijing, China; Table 1). Partial genes of ferlavirus, reovirus, and adenovirus were amplified by nested PCR.2,20,21 Sunshine virus and nidovirus were detected by conventional PCR.7,19 In order to ensure true positives, samples were further screened for ferlaviruses by another conventional PCR method.7,11
PCR was performed in a final reaction volume of 25 μL, comprised of 12.5 µL of 2× DNA polymerase master mix (CW Bio), 0.4 µM (1 µL) of each primer, 2 µL of DNA or cDNA template, and 8.5 µL of RNase-free water (CW Bio). The PCR procedure was as follows: initialization at 94°C for 10 min; denaturation at 94°C for 30 s; annealing for 30 s at 45°C, 45°C, 47°C, and 46°C for ferlavirus, sunshine virus, reovirus, and adenovirus, respectively; extension at 72°C for 60 s; 35 cycles of denaturation, annealing, and extension steps; and final elongation at 72°C for 5 min. A touchdown PCR was performed on nidovirus as follows: 9 touchdown cycles at 95°C for 10 s; 57.5°C for 30 s (–1°C per cycle); 72°C for 50 s; 26 cycles at 95°C for 10 s; 49.5°C for 30 s; 72°C for 50 s; and a final extension at 72°C for 7 min. 13
Obtained PCR products were separated on 1.5% agarose gel and collected (Gel extraction kit; CW Bio) under ultraviolet visualization (ImageMaster VDS-CL system; Bio-Rad, Hercules, CA). Purified PCR segments were inserted into a pUC-T plasmid vector (pUC-T quick ligation kit; CW Bio) and transfected into heat-shocked Escherichia coli DH5α competent cells (CW Bio) by ligating at 42°C for 45 s; these transfected cells were seeded on selective agar plates containing ampicillin. Positive clones were randomly selected and purified (Pure plasmid mini kit; CW Bio), and then sequenced by the Beijing Genomics Institute.
Sequences were edited, assembled, and analyzed using the DNASTAR Lasergene package (https://www.dnastar.com/software/lasergene/). Sequences were also compared with online data available in GenBank (https://www.ncbi.nih.gov) using TBLASTn and BLASTx analyses. Multiple alignments of homologous nucleotide and deduced amino acid sequences were evaluated by the ClustalW method (https://www.genome.jp/tools-bin/clustalw). Phylogenetic relationships of the sequences were further described by the maximum-likelihood and neighbor-joining methods in MEGA v.5.0 (https://www.megasoftware.net/). The bootstrap analysis was replicated 1,000 times.
We found ferlavirus infection to be common in farmed snakes with respiratory disease in Guangxi; 70 of 104 snakes tested were infected (Table 2). Using the nested PCR method (PCR I), ferlaviruses were detected in 58 of 104 snakes. However, 70 of 104 snakes were found to be ferlavirus positive after a conventional PCR (PCR II), suggesting that PCR II is more sensitive for ferlavirus detection than PCR I, which is consistent with the findings of a previous report. 11 The 70 ferlavirus-positive snakes included 49 oriental rat snakes, 19 cobras, and 2 king rat snakes, which were obtained from 34 of the 52 snake farms in Guangxi Province (Table 2). The positive detection rate of snake samples and farms was generally ≥ 50%. No adenovirus, reovirus, sunshine virus, or nidovirus was detected in any of the samples tested.
We obtained 96 bacterial isolates from the lung, liver, and heart of 77 of 92 (84%) snakes, and the same bacteria isolated from different organs of the same snake were counted as one isolate. Bacteria were isolated from 51 of 66 (77%) ferlavirus-positive snakes and from 26 of 26 (100%) ferlavirus-negative snakes. The bacterial isolates included 18 Salmonella, 16 Proteus, 11 Pseudomonas, 7 Enterococcus, 7 Staphylococcus, 6 Providencia, 5 Escherichia coli, 5 Stenotrophomonas, 4 Bordetella, 4 Myroides, 3 Alcaligenes, 3 Morganella, 2 Aeromonas, 2 Chryseobacterium, 2 Shewanella, and 1 Cronobacter species. Most of these bacteria are normal gastrointestinal flora or environmental microbes that can cause disease when animals are in poor health. 18 Our high bacterial isolation rate from snakes with respiratory disease suggests that bacteria could be a primary factor in respiratory disease, at least in ferlavirus-negative snakes. However, 23% of ferlavirus-positive snakes were bacteria negative, suggesting that ferlavirus may have been the most likely primary pathogen in those cases of respiratory disease. Immunosuppression may be induced by ferlavirus, resulting in secondary bacterial infection that complicates disease expression.9,17
Other pathogens, including mycoplasma, fungi, and arenavirus, which were not detected in our study, may also be involved. Moreover, poor husbandry conditions are probably important triggers for the development of respiratory disease. 17
The nucleotide sequences of the L gene in ferlaviruses are highly conserved and serve as indicators for virus phylogeny and classification.2,16 The amplicon acquired using PCR II was149 bp, which is relatively short and not suitable for phylogenetic studies. 11 Therefore, we used 566 bp of L gene sequences obtained by PCR I for sequence analysis and describing the phylogenetic tree. Sequence analysis of 58 PCR I products revealed that ferlaviruses collected from different snakes from the same farm were identical. We submitted 20 different partial L gene sequences to GenBank (accessions MK368615–MK368634), from the 27 ferlavirus-positive farms.
Homology analysis (Table 3) and the phylogenetic tree (Fig. 2) revealed that the 20 different partial L gene ferlavirus sequences obtained from Guangxi could be divided into 3 distinct genogroups, which were consistent with groups A–C of previous reports.14,15 In group A, the 8 strains from Guangxi and the known strains Fer-de-lance, Gono-GER85, Snake ATCC-VR-1408, Xeno-USA99, ElaGut-91, Crotx-96, AnaPV, and Dasy-GER00, clustered together and formed several clades. The Guangxi strains formed a separate clade with nucleotide homologies of 98.6–99.8% and nucleotide homologies of only 85.3–90.0% with the GenBank strains.
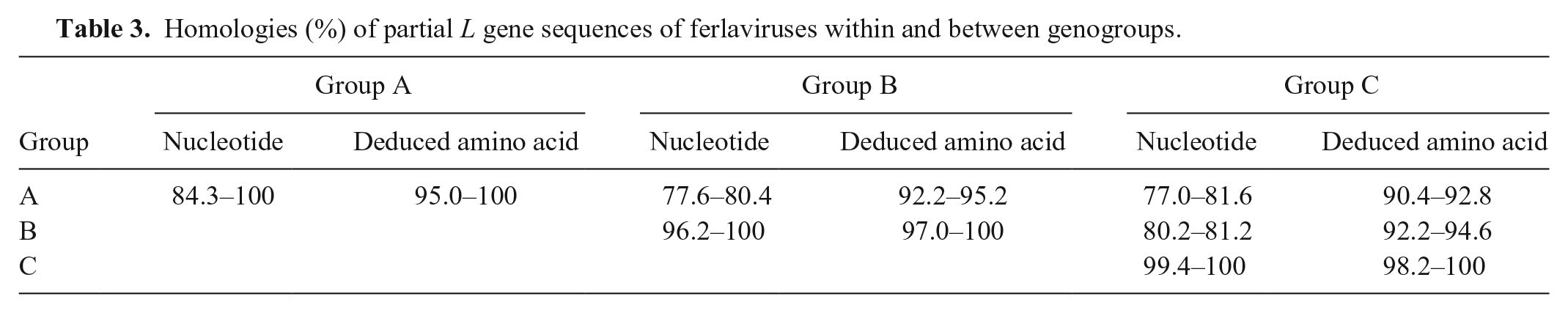
Homologies (%) of partial L gene sequences of ferlaviruses within and between genogroups.
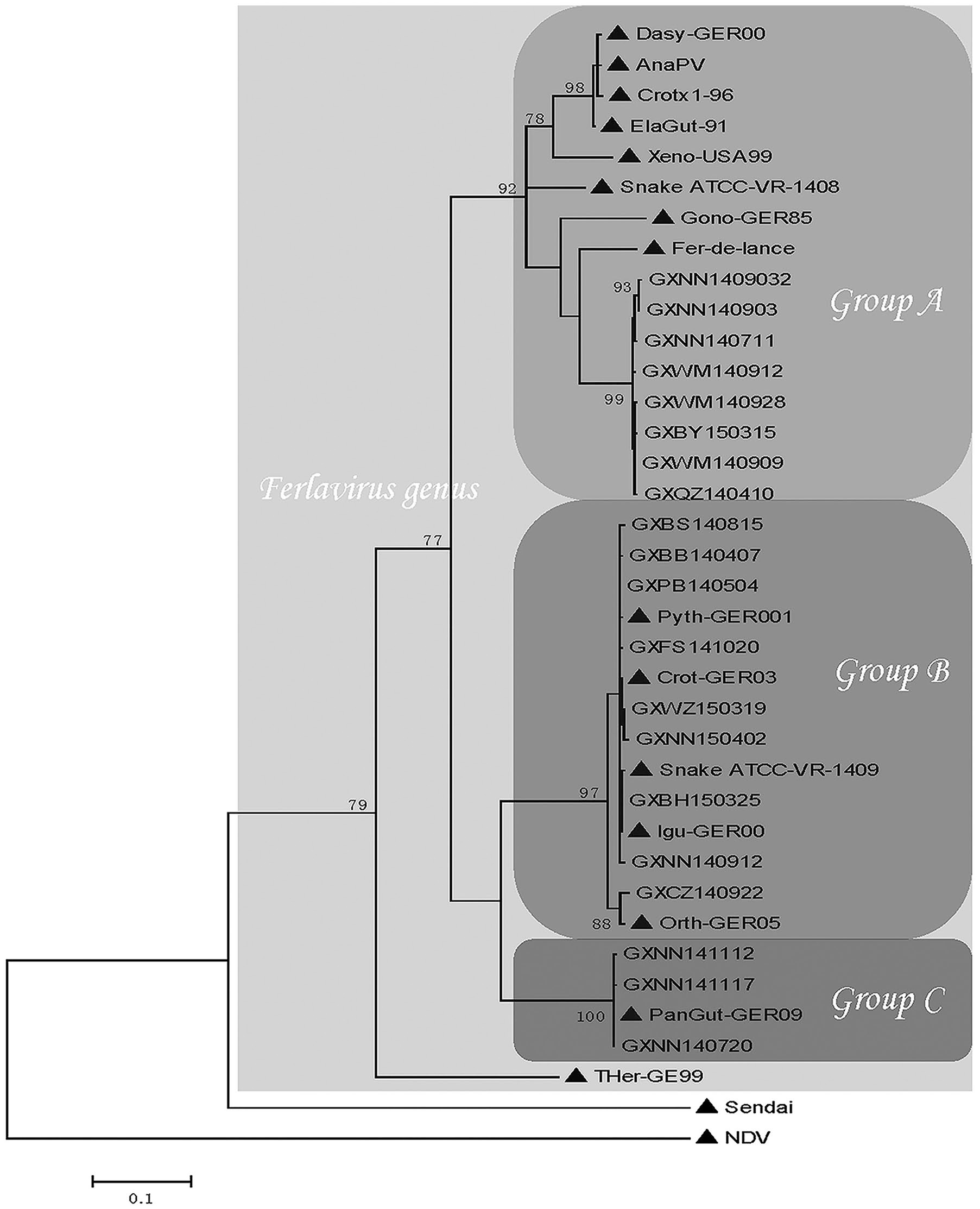
Phylogenetic distance tree based on partial L gene sequences (501 nt) of ferlaviruses in Guangxi Province, China and published ferlaviruses (marked with ▲) Fer-de-lance (NC005084), Crotx1-96 (AF349405), Dasy-GER00 (GQ277613), Xeno-USA99 (GQ277614), ElaGut-91 (AF349408), Snake ATCC-VR-1408 (AF28604), AnaPV (KY745892), Snake ATCC-VR-1409 (AF286043), Orth-GER05 (GQ277616), Crot-GER03 (GQ277611), Pyth-GER001 (GQ277612), lgu-GER00 (GQ277617), PanGut-GER09 (HQ148084), and THer GER (GQ277615). Corresponding regions of Newcastle disease virus (NDV; AF375823) and Sendai virus (NC001552) were used as outgroups in the calculations.
Group B consisted of 9 strains from Guangxi and 5 known GenBank strains, including Grot-GER03, Pyth-GER001, lgu-GER00, Snake ATCC-VR-1409, and Orth-GER05. The 3 strains from Guangxi and the published PanGut-GER009 strain clustered closely into group C. Both the nucleotide and deduced amino acid homology values were ≥ 96.2% within groups B and C. Within group B, strains GXCZ140922 and Orth-GER05 clustered into a separate branch with a nucleotide homology of 96.2−97.3%, and amino acid homologies of 97.0−98.1% with other strains in group B. These 2 strains may be the result of mutations. Compared to groups B and C, large variations within group A suggest that ferlaviruses in group A are multiphyletic and could be further divided into several distinct subgroups. 10
The classification of genus Ferlavirus found in Guangxi does not show a geographic difference from Ferlavirus found in Europe and the United States.1,14,16 We found that the same group of ferlaviruses can be present in multiple locations of Guangxi, and different groups may coexist within the same location. For example, the GXPB140504 (Pubei), GXBS140815 (Baise), GXNN140912 (Nanning), GXMS141112 (Mashan), GXWM141202 (Wuming), GXWZ150319 (Wuzhou), and GXBH150325 (Beihai) strains were clustered into group B with a deduced amino acid homology of 100%. All 3 groups of ferlaviruses were found in snakes from Nanning, and both group A and B were found in Qinzhou. Based on our results, the epidemic patterns of ferlaviruses in Guangxi are both diverse and complex.
Our findings confirm that farmed snakes in Guangxi are at risk of ferlavirus infection. To date, no drugs or vaccines are available to effectively prevent or treat this virus infection. Preventive control measures, such as pathogen detection before introducing new snakes, regular disinfection of snake farms, and providing good animal welfare, are suggested as effective means for protecting snakes from ferlavirus infection.
Footnotes
Acknowledgements
We thank the Institute for Poultry Science and Health of Guangxi University for providing the reovirus and adenovirus isolates. We also thank Shi-wen Feng, Li-li Liao, Min-ling Lin, and Rui-jun Li from Guangxi University for their technical assistance. We thank LetPub (![]() ) for their linguistic assistance during preparation of this manuscript.
) for their linguistic assistance during preparation of this manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Civic Science and Technology Project of Nanning (20163355) and Application Popularization Project of Department of Forestry of Guangxi Zhuang Autonomous Region.