
Abstract
Clostridium perfringens type D epsilon toxin (EXT) causes an important neurologic disorder of sheep, goats and, rarely, cattle. The disease can occur in peracute, acute, subacute, and chronic forms. High circulating levels of ETX produce vasculocentric brain lesions, in which microvascular endothelial injury results in diagnostically useful perivascular and intramural extravasations of plasma protein, especially in sheep, and less frequently in goats. With lower toxin doses, a more protracted clinical course tends to occur, particularly in sheep, leading to focal, bilaterally symmetrical, necrotic foci in certain brain regions. Although these morphologic features usually permit the diagnostic pathologist to make a definitive etiologic diagnosis, there are many aspects of the pathogenesis of these cerebral lesions that are not completely understood. ETX has also been shown to produce microvascular damage in the retina of rats, resulting in severe, diffuse vasogenic edema, similar to that found in brains exposed to this neurotoxin. The pathoclisis and vascular theories offer alternative explanations of the differential susceptibility of different brain regions to the same neurotoxic insult.
Keywords
Clostridium perfringens type D enterotoxemia occurs commonly in sheep, particularly lambs, and is of worldwide distribution.28,32,37 It is also an important disease of goats and, less frequently, cattle.35,36 Type D strains are characterized by the production of 2 typing toxins, namely alpha (CPA) and epsilon (ETX). The latter is, however, the main virulence factor of this microorganism.31,32,37
Some ruminants harbor C. perfringens type D in their small intestine, but bacterial numbers are usually small, and low levels of ETX production are relatively harmless. However, when the microbial balance in the gut is disrupted, frequently by a sudden overload of ingested carbohydrates, these saccharolytic bacteria proliferate rapidly and elaborate large amounts of ETX. When high luminal concentrations of this toxin are produced, enough ETX can be absorbed into the bloodstream to produce severe neurologic and respiratory disturbance. 38
The clinical course produced by ETX spans a continuum. When large amounts of toxin enter the circulation, especially in lambs, there is sometimes a peracute course, with sudden death and no premonitory signs. Less acutely intoxicated animals have severe convulsions, which can lead to coma and, most frequently, death. 30 By contrast, when ETX doses are lower, or animals are partially immune, there is subacute or chronic intoxication with a more protracted clinical course. These sheep and goats have neurologic signs that include blindness, aimless wandering, ataxia, bruxism, head pressing, nystagmus, opisthotonos, and paddling convulsions. 14 Goats with subacute or chronic intoxication also have diarrhea; intestinal clinical signs are not usually seen in sheep with this disease. 37
In sheep and goats, gross lesions in peracute, acute, and subacute cases can include pulmonary edema, hydropericardium, ascites, and hydrothorax, with or without fibrin strands in body cavities. Occasionally, no gross lesions are observed at autopsy; herniation of the cerebellar vermis (Fig. 1) can be seen in a few acute and subacute sheep cases. 37 In addition, goats with acute and subacute disease have necrohemorrhagic colitis or enterocolitis.
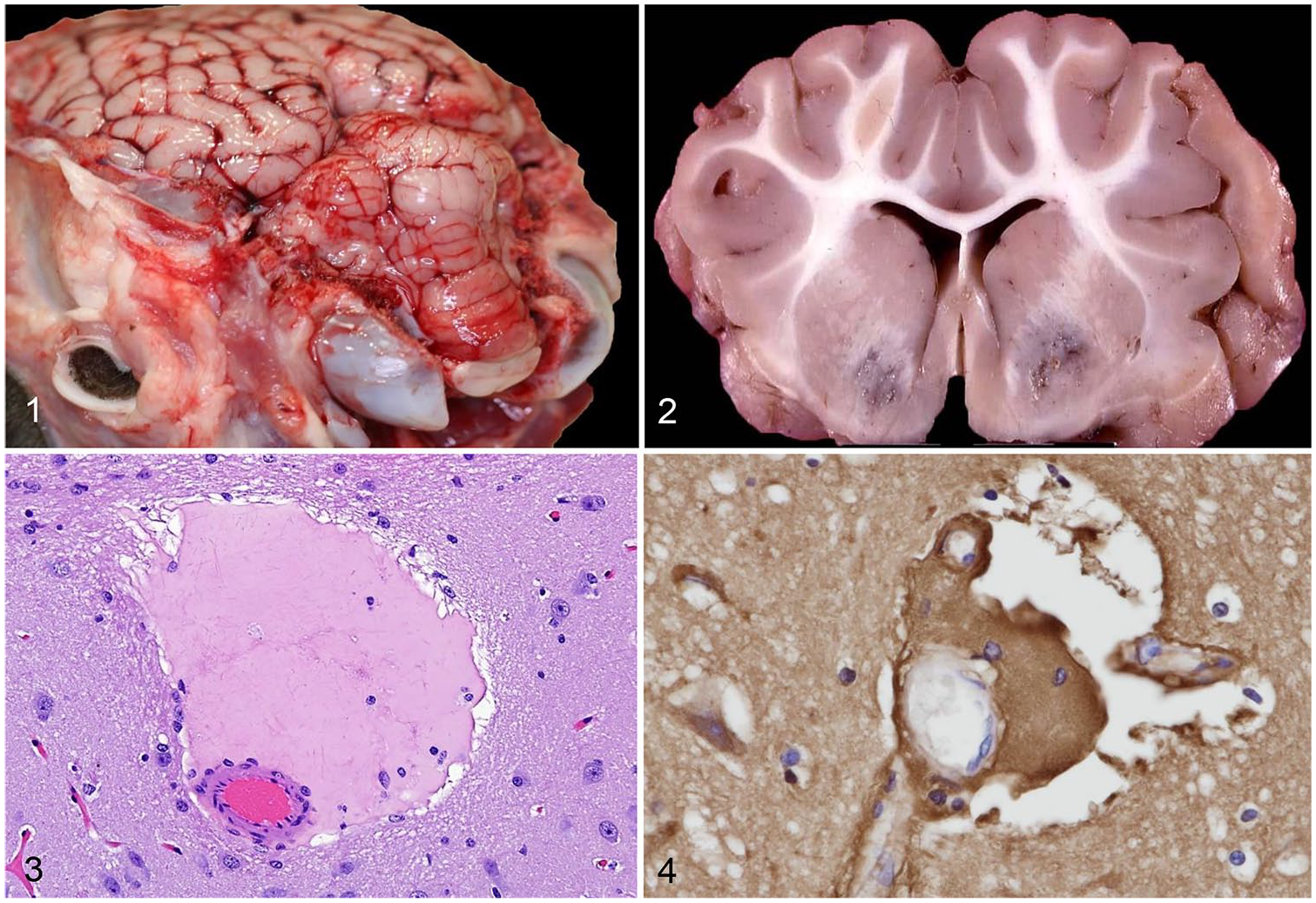
Clostridium perfringens type D enterotoxemia in sheep.
In subacutely and chronically intoxicated sheep, following a more prolonged clinical course, bilaterally symmetrical foci of malacia (Fig. 2) can occur in certain brain regions of predilection, including the basal ganglia, thalamus, internal capsule, midbrain, medulla oblongata, and cerebellar peduncles. This neuropathologic change is termed focal symmetrical encephalomalacia (FSE). 16 This lesion has been described, albeit infrequently, in goats and cattle, but conclusive evidence that it is produced by ETX is lacking.
In peracutely, acutely, and subacutely intoxicated sheep, and less frequently in goats, the finding of perivascular and vascular intramural lakes of plasma protein–rich fluid (Fig. 3) in the brain is typical of type D disease.12,13,15,29 This edema contains a high concentration of albumin (Fig. 4). In regions of predilection for blood–brain barrier breakdown, this vasogenic edema is more commonly found in white than gray matter, particularly that of the cerebral cortex, corpus striatum, thalamus, midbrain, medulla, and cerebellum. 12 The sparse cellularity and larger extracellular spaces in white matter permit greater accumulation, and spread, of vasogenic edema fluid. 26
These vascular lesions reflect the fact that the microvascular endothelium appears to be the principal target in ETX neurotoxicity. 9 The primacy of microvascular damage in the pathogenesis of ETX neurotoxicity is also supported by the finding that this toxin produces a rapid and dose-dependent cytotoxic effect on cerebral microvascular endothelial cells in vitro. 21
For the diagnostic anatomic pathologist, the vasculocentric lesions found in peracutely, acutely, and subacutely intoxicated ruminants are sufficient for a diagnosis of type D enterotoxemia, abetted by ETX detection in intestinal contents. Other ancillary tests such as intestinal smears for proliferating gram-positive rods and detection of glucose in urine are supportive, but not confirmatory, of a diagnosis of enterotoxemia in sheep. 36 Similarly, FSE is pathognomonic for the more chronic manifestation of ETX intoxication in sheep. 36 However, although it is usually possible to make a definitive diagnosis using these morphologic criteria, there are many aspects of the development of ETX-induced brain lesions that are not completely understood.
ETX is a member of the aerolysin family of pore-forming toxins and causes endothelial damage after binding to specific receptors. 34 This endothelial injury can be prevented, albeit transiently, by competitive inhibition of ETX receptors from prior administration of the much less toxic prototoxin.3,27 One domain of ETX binds to endothelial receptors, where it uses lipid rafts and caveolin to oligomerize into a pre-pore on the surface of the cell membrane, while another domain inserts into the membrane lipid bilayer and forms an active pore. Pore formation leads to a rapid decline in cytoplasmic potassium levels, and an influx of sodium and chloride ions, depleting ATP; this results in endothelial cell necrosis. 34
In ETX-intoxicated ruminants, even when the brain is exposed to large doses of circulating toxin, only a small number of microvessels have endothelial damage sufficient to result in protein-rich intramural and/or perivascular edema.12,29 Although this endothelial damage can be detected by light microscopy, it is best seen at the ultrastructural level. There is initially endothelial swelling, with loss of cytoplasmic organelles and blebbing of the luminal surface. The cytoplasm is later reduced to an attenuated electron-dense band with nuclear pyknosis.7,13 To date, there is no satisfactory explanation for the inconsistency, both in frequency and neuroanatomic distribution, of these vasculocentric lesions between individual cases. Moreover, in the subacute, and more chronic, form of ETX intoxication, manifest as FSE, it is not known why focal, and bilaterally symmetrical, necrosis occurs preferentially in certain neuroanatomic sites.
It is possible that only a subset of cerebral blood vessels possesses endothelial receptor sites and thus sustains toxin damage. Alternatively, given that ETX neurotoxicity appears to be expressed in a dose- and time-dependent manner, these factors could determine which microvessels in selected brain areas are exposed to sufficient toxin to cause endothelial damage. 9 Experimentally, ETX has been shown to cause either selective or more widespread neural injury, depending on the dose of circulating toxin and the time interval between toxin administration and death. The resulting brain lesions are quite variable in both number and regional distribution. In general, higher doses of ETX tend to expand the distribution of brain lesions, although still favoring certain vulnerable areas, whereas lesions in subacutely intoxicated animals exposed to lower doses of ETX are smaller, tend to be well-circumscribed, and are restricted to fewer susceptible brain regions. Moreover, as the time from toxin administration to death increases, the distribution of brain lesions tends to become more widespread. 9 It has also been shown in mice given ETX that multiple sublethal doses of toxin produce lesions in a wider range of neuroanatomic sites than a single dose. 7
In ETX-intoxicated goats, it has been speculated that the more frequent occurrence of intramural than perivascular edema is a reflection of lower doses of toxin being absorbed from the intestine in this species compared to sheep, resulting in less severe cerebral microvascular injury. 29 Another possible reason for the lack of uniformity in microvascular damage produced in the brain by ETX is the recognition that cerebral endothelial cells constitute a heterogeneous population, this variability being attributed not only to the functional diversity between different brain regions, but also among capillaries and venules, and even at the single cell level within individual capillaries. 40
In FSE, and other animal neurologic disorders in which bilaterally symmetrical necrotic foci are found, such as equine nigropallidal encephalomalacia and focal symmetrical poliomyelomalacia of sheep, cattle, and pigs, 4 the gross and microscopic nature of this necrosis is similar, but the differing neuroanatomic pattern of distribution of these necrotic lesions can be diagnostically useful.
It is a well-recognized phenomenon in neuropathology that, although an insult is usually delivered to the entire brain, only a subset of neurons in certain regions are damaged, leading to their dysfunction or death. This concept is designated selective vulnerability. 1 Although the mechanisms of selective vulnerability are, for most neurologic disorders, unknown, a given insult nevertheless generally injures specific brain regions and the resulting lesion tends to be characteristic of that particular disease. 23 However, selective vulnerability is due, in part at least, to the fact that the brain is the most inhomogeneous organ in the body, with different brain regions varying structurally and serving very different functions. The brain is also extraordinarily complex, being comprised of highly interconnected networks involving numerous nuclei. 1 Two broad explanations have been proposed for selective vulnerability in the brain. The pathoclisis theory postulates that different neuronal molecular profiles between different brain regions account for the differential susceptibility to a given insult, together with varying reactions to changes in the perineuronal microenvironment. By contrast, the vascular theory posits that the various vascular patterns between different neuroanatomic sites are an important factor contributing to different vulnerabilities. 1 However, although cogent explanations for selective vulnerability are still lacking for most neurologic disorders, there are exceptions. After transient global ischemia–hypoxia, for example, there is excessive release of the neurotransmitter glutamate in regions in which excitotoxic receptors are abundant, such as the CA1 region of the hippocampus and cerebellar Purkinje cells. This leads to excitotoxic neuronal death. 2 Another example of a known susceptibility of a specific brain region is the propensity of carbon monoxide to cause histotoxic hypoxic injury to the globus pallidus and substantia nigra given its high affinity for the cytochrome heme iron in these areas. 18
The development of FSE lesions could reflect higher susceptibility to ETX of the neural parenchyma or the vasculature in certain brain regions, but the reason for this vulnerability remains elusive. 41 It is possible that, after gaining access to the brain by damaging the microvasculature, ETX could have a differential cytotoxic action on specific neuroanatomic sites. 8 Microvascular injury produced by ETX could also cause thrombosis, abetted by vascular compression in these smaller vessels from severe astrocytic end-feet swelling resulting from increased vascular permeability. The resulting localized (focal) failure of perfusion could then cause ischemic–hypoxic damage. However, the development of the large necrotic foci found in FSE would require either obstruction of a large area of the microvascular bed or occlusion of perforating end-arteries, and neither of these vascular events has been demonstrated to date. 9
Although there have been several studies examining the neurotoxicity of ETX in vitro, and in laboratory rodents,3,5–7 it has often been difficult to relate these experimental findings to the pattern of lesions found in naturally occurring ruminant cases. ETX has been shown to cause excessive release of glutamate from pre-synaptic, glutamate-rich sites in rat and mouse hippocampi, leading to post-synaptic dendritic injury and pyramidal cell death.24,25 ETX also binds to the soma and dendrites of glutamatergic mouse cerebellar granule cells. 20 Moreover, ETX has been reported to damage dopaminergic areas of the murine brain. 27 This toxin has also been shown to bind to, and kill, oligodendrocytes in a dose- and time-dependent manner in brain cultures, reportedly leading to demyelination. 19 Myelin binding of ETX has also been described. 5 It has recently been found that some multiple sclerosis patients have serologic evidence of exposure to ETX and it has been suggested that the toxin may play a role in the development of this human demyelinating disorder. 39 However, this human disease is very different from the naturally occurring disease produced by ETX in domestic livestock and laboratory rodent models.
In addition to localizing in the brain, circulating ETX accumulates in the eye. 33 The blood–retinal barrier (BRB) resembles the blood–brain barrier (BBB) in most important respects, 17 and it has been shown, in acutely intoxicated rats, that ETX injures the BRB in a manner resembling that of the BBB. This results in severe, diffuse vasogenic retinal edema. 11 Using endogenous albumin as a surrogate immunohistochemical marker of increased vascular permeability, there is diffuse albumin immunopositivity in all retinal layers. More robustly, albumin floods the injured vascular endothelium and accumulates in perivascular sites. Ultrastructural examination reveals severe endothelial damage, which is characterized by marked attenuation and increased electron density of endothelial cells, resembling that found in cerebral endothelial cells of sheep and mice exposed to ETX.7,13 ETX-induced microvascular injury in the rat retina (and brain) also correlates with loss of endothelial barrier antigen (EBA) immunoreactivity, EBA being a marker of an intact BRB (and BBB) in this species.10,22 This EBA finding confirms that vascular disruption, and the ensuing generalized, high-protein edema, is the essential retinal lesion produced by ETX.
The vasculocentric, and focally, bilaterally symmetrical, necrotic lesions found in type D disease in the brains of ruminant livestock are generally sufficiently characteristic for the diagnostic pathologist to make an etiologic diagnosis. However, there are many aspects of the pathogenesis of these morphologic changes that are, as yet, unexplained. The damaging effect of ETX on the retinal vasculature, which has recently been demonstrated in rats, also warrants further investigation, particularly as this ocular lesion is likely to contribute to a visual deficit, and even blindness, in affected ruminants.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.