
Abstract
Mycoplasma hyopneumoniae causes highly contagious swine enzootic pneumonia worldwide. It has been reported that highly diversified M. hyopneumoniae strains exist in different parts of the world. We found p146 gene sequencing analysis, an affordable and simple-to-perform typing method, to be specific and highly sensitive when applied to the molecular typing of 113 M. hyopneumoniae–positive clinical samples directly without culture. The samples were submitted to the Animal Health Laboratory at the University of Guelph (Ontario, Canada) during 2009–2017 from 40 different geographic areas in Ontario. Using a previously described criterion of grouping strains with < 4-bp differences into the same molecular type (p146 type), the 113 clinical samples clustered into 19 p146 genotypes. Dominant types were found in 2016 and 2017 only, indicating that highly diversified M. hyopneumoniae strains existed in Ontario. Some strains from the same geographic location but different years had the same sequence types, indicating that the same strain types circulate persistently in the field. Different p146 genotypes were also identified from similar geographic locations, indicating that either M. hyopneumoniae strains are prone to mutation or that multiple strains can infect the same or nearby swine production units.
Mycoplasma hyopneumoniae is a highly contagious cell-wall–free bacterium known to cause porcine enzootic pneumonia. The bacterium causes significant growth weight reduction in young grower pigs and increases medication use, both of which effects cause significant economic losses. 1 Genetic typing of this pathogen can be useful in M. hyopneumoniae disease diagnosis and epidemiologic analysis. M. hyopneumoniae multilocus variable-number tandem repeat analysis (MLVA) and multilocus sequence typing (MLST) have been described.5,9 However, these methods are complicated to run, somewhat difficult to interpret, and not affordable for routine testing. The polyserine repeat encoding region of the p146 gene is highly polymorphic and useful for M. hyopneumoniae typing directly from field samples without culture. 4 It has been described that p146 gene typing could complement both MLST and MLVA methods in order to refine more closely the genetic relationships of M. hyopneumoniae, and that p146 gene sequencing typing is adequate for the resolution of genetic relationships within MLST groups. 3 Our objective was to use p146 gene direct sequencing to characterize M. hyopneumoniae–positive samples collected in the past 10 y from pigs in Ontario, Canada.
We selected randomly 113 clinical samples positive by a M. hyopneumoniae quantitative PCR (qPCR; Mhp183 qPCR) 8 to be tested by p146 gene PCR sequencing. The samples were submitted to the Animal Health Laboratory (AHL) at the University of Guelph (Guelph, Ontario, Canada) from 2009 to 2017. Sample types included 82 lung tissues, 28 oral fluids, and 1 swab sample, from 40 different geographic locations in Ontario as defined by Canadian postal codes. In addition, type strain M. hyopneumoniae J 7 and well-characterized reference M. hyopneumoniae strain 232 6 were also included.
Within 48 h of receiving samples, DNA was extracted from oral fluids (300 µL each) or swab samples (100 µL) using the MagMax Express-96 automated nucleic acid extraction system (Thermo Fisher, Burlington, ON). The type and reference strain broth cultures (100 µL) were handled in the same manner. DNA from lung tissue was extracted (DNeasy blood and tissue kit; Qiagen, Toronto, ON) following the manufacturer’s instructions. DNA extracts were either stored at 4°C (same-day testing) or −20°C or −80°C (later testing).
The primers and protocol of the p146 gene sequencing typing assay were as described previously, with a final concentration of 3 mM MgCl2, 0.2 mM dNTPs, 0.2 µM primers (Mhp684F: 5′-ACTCCAAGACGAAGATCTTGACTATCAAAT-3′; Mhp684R: 5′-CCGATATTAGAACTTGCAAGATAAAGC-3′),4,9 and 0.75 units of Taq Gold (Thermo Fisher) in a 25-µL reaction volume. Cycling parameters were 10 cycles of 94°C for 30 s, 63°C for 30 s (with a temperature decrease of 1°C per cycle), 69°C for 75 s, followed by 40 cycles of 94°C for 30 s, 53°C for 30 s, 69°C for 75 s, and a final extension step of 5 s at 69°C. Products were held at 4°C for a maximum of 48 h before further analysis. The p146 gene PCR products were purified and sequenced using the same primers in 2 directions (Applied Biosystems 3730 DNA analyzer; Thermo Fisher). To verify the sensitivity of the p146 gene PCR, it was compared with the Mhp183 qPCR, a PCR commonly used to detect M. hyopneumoniae infection. 8
The p146 PCR products were sequenced in both directions, and the computer program DNASTAR (SeqMan Pro, Lasergene 14; DNASTAR, Madison, WI) was used to assemble the sequences into a consensus sequence. The primer sequences were deleted, and consensus sequences [184 bp ~ 229 bp] were further analyzed (MegAlign, Lasergene 14, DNASTAR) using the ClustalW method with the following multiple alignment parameters: gap penalty 10.0, gap length penalty 0.2, delay divergent sequences 30%, DNA transition weight 0.5; and pairwise alignment parameters: gap penalty 10 and gap length 0.1. A total of 19 clusters were observed from the phylogenetic tree formed from MegAlign (Fig. 1). SeqMan Pro was used to compare the consensus within each cluster and subcluster from the phylogenetic tree. Sequences with < 4-bp difference (~2% of the sequence) were automatically grouped into the same cluster as the same molecular type (p146 type). Those sequences with 1- to 2-bp differences within the same molecular type were grouped into a subcluster as a subtype.
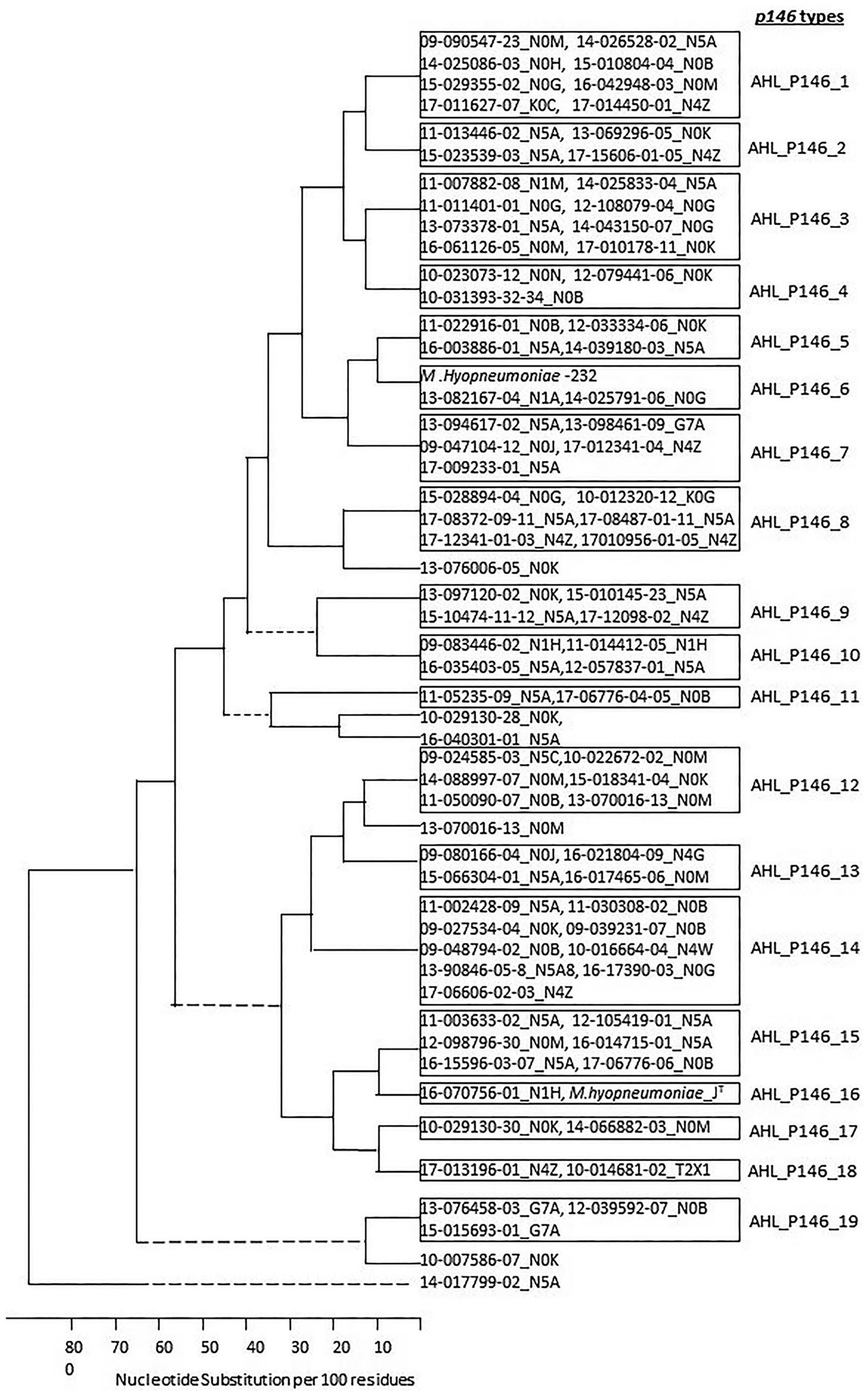
The phylogenetic tree of the AHL_p146 types of 113 Mycoplasma hyopneumoniae field strains identified at the Animal Health Laboratory. The first part of the strain name indicates the year of isolation and case number. The second part of the strain name is the postal code of the location from which the samples were obtained. A new p146 type was assigned only when the type group consisted of >2 sequences.
The p146 sequence PCR was specific and highly sensitive. The p146 gene PCR was only positive with 3 M. hyopneumoniae pure cultures and 110 M. hyopneumoniae–positive samples detected by Mhp183 qPCR. There was no false-positive result with any non–M. hyopneumoniae mycoplasma or any of the other bacteria commonly found in pig samples including Trueperella pyogenes (AHL strain 104), Actinobacillus suis (AHL strain 110), Clostridium perfringens (NCTC 3110), Enterobacter cloacae (AHL strain 80), Escherichia coli (ATCC 25922), Erysipelothrix rhusiopathiae (AHL strain 111), Klebsiella pneumoniae (AHL strain 82), Mycoplasma arginini (NCTC 10129T), M. hyorhinis (MH31), M. hyosynoviae (S16), Pseudomonas aeruginosa (AHL strain 347), Salmonella Enteritidis (AHL strain 232), Salmonella Typhimurium (AHL strain 322), Staphylococcus aureus (AHL strain 87), and Streptococcus suis (AHL strain 109). All of the non-target organisms were either AHL field isolates, confirmed by phenotypic and/or molecular identification, or were reference or type strains. The detection limit for the longer cycles of p146 gene PCR (50 cycles) was 0.2–2 CFU per reaction; the detection limit for the Mhp183 qPCR (45 cycles) was 2 CFU of M. hyopneumoniae pure culture per PCR reaction. Both PCR assays could detect M. hyopneumoniae in undiluted to a 105 dilution of a DNA extract of a field sample. The PCR target and the primers and probe used in the Mhp183 assay were previously found to be able to identify multiple isolates. 8 Given that the p146 gene PCR was able to amplify all of the Mhp183 PCR–positive samples, it can potentially be used for both detection and typing of M. hyopneumoniae directly from samples without culture.
From 113 M. hyopneumoniae–positive samples collected during 2009–2017 from 40 different geographic areas in Ontario, we identified 19 p146 types of M. hyopneumoniae that included 4 more subtypes using the MegAlign default cluster criteria, which are similar to the cluster designation criteria described previously (Fig. 1). 4 Under these criteria, a sequence with ≥ 4-bp difference is assigned to a new p146 type, and a sequence ≤ 2-bp differences within the same p146 type is assigned to a new subtype. 4 Our study revealed that a large number of M. hyopneumoniae variants have circulated on Ontario farms. Other studies have demonstrated that multiple M. hyopneumoniae variants were circulating in swine by using MLST, MLVA typing, and p146 gene sequence typing.2,4,5,9 Although 2 of the AHL M. hyopneumoniae samples were grouped with the non-Canadian origin M. hyopneumoniae 232 (AHL_p146_6) and type strain J (AHL_p146_16), respectively, most (95%) of the AHL M. hyopneumoniae belong to novel p146 types. The sequences of these novel types, alleles 26, 32, 33, and 137-157 of locus p146, are available from the M. hyopneumoniae PubMLST p146 database (https://pubmlst.org/bigsdb?db=pubmlst_mhyopneumoniae_seqdef&page=locusInfo&locus=p146). The finding that a large number of different p146 types circulate in Ontario indicates that mutation may occur relatively frequently in M. hyopneumoniae.
Two dominant strains were found, one in 2017 and one in 2016. In 2017, 18 of 31 samples from 3 different geographic locations were grouped into 3 closely related clusters, with 15 samples being AHL_p146_8. The AHL_p146_8 cluster included only 2 samples collected in other years, one each from 2015 and 2010. The remaining 17 of the 2017 samples were sporadically clustered in other AHL p146 types. Similarly, 5 of 12 samples collected in 2016 from 2 different geographic locations were grouped into closely related clusters, with 4 being AHL_p146_15, and 1 being the closely related AHL_p14_16. Interestingly, the 2016 dominant strains (AHL_p146_15 and _16) are closely related to strain M. hyopneumoniae J, which belongs to AHL_p146_16. Predominant strains were not found from 2009 to 2015. The occurrence of different dominant strains in different years has not been reported previously, to our knowledge.
Our study showed that a specific geographic area can have dominant strains, although these strains can be found in areas as far as 100 km apart and can be found in different years. Most samples from postal code N4Z belonged to either AHL_p146_8 (7/16) or AHL_p146_2 (3/16) and were detected only in 2017. Most samples of type AHL_p146_15 were found in postal code N5A (3 km north of N4Z) and were collected in 2011, 2012, and 2016. In addition to postal code N5A, AHL_p146_15 was found in postal code N0B (52 km northeast of N5A) in 2017 and N0M (104 km southwest of N5A) in 2012. This indicates that the AHL_p146_15 genotype has circulated persistently and spread widely in the field. Similarly, AHL_p146_1 was found in 2009 and 2014–2017, and AHL_p146_3 was found from 2011 to 2017 from different areas. The fact that some strains of the same p146 types were found from the same geographic location but different years indicates that the same strain types are persistent and are widely circulating in the field. In contrast, different p146 genotypes were also identified from similar geographic locations, indicating that either M. hyopneumoniae strains are prone to mutation or that multiple strains can infect the same or nearby swine production units. A similar high diversity of M. hyopneumoniae has been reported in Switzerland.4,5 In addition, the same Swiss studies found that farms in close geographic or operative contact generally seemed to be affected by the same strains, and single pig farms harbored the same farm-specific strain,4,5 a situation that was not obvious in our study in Ontario.
There were not enough clinical data to analyze the link of strain types with clinical presentation in our study. An affordable typing assay such as p146 gene sequencing will allow further investigation of the pathogenesis of disease caused by different genotypes of M. hyopneumoniae.
After the completion of the analysis of historical samples, we received 7 samples from 3 farms near each other to investigate if the M. hyopneumoniae infection had spread from one to the other. Among the 7 samples, 3 samples from the same farm were classified as AHL_p146_4; 3 samples from another farm were identified as p146_86 type registered previously in the PubMLST database, different from any historical types indentified at the AHL. The remaining sample from the third farm had a 9-bp plus 57-bp insertion in the p146 gene, resulting in 22 additional serine repeats, which made the PCR product sequence (280 bp) much longer than others (213 bp). The sequence was classified and deposited to the PubMLST database as p146_157. Based on these typing result differences, it would seem that the infection did not spread among the 3 farms.
Footnotes
Acknowledgements
We thank Dr. Chris Minion for his generous gift of well-characterized M. hyopneumoniae 232.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was financially supported by the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA) Disease Surveillance Program, managed by the Animal Health Laboratory.