
Abstract
We compared the effectiveness of various methods for the identification of Staphylococcus spp. other than S. aureus isolated from intramammary infections of cows on 3 dairy farms in Lower Silesia, Poland. A total of 131 isolates belonging to 18 Staphylococcus species were identified by sequence analysis of the 16S rRNA and dnaJ genes, as well using a commercial identification system (ID 32 STAPH; bioMérieux) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS; Bruker Daltonics). Sequencing of the 16S rRNA gene was found to have low discriminatory value because only 43% of isolates were recognized unequivocally. Much better results were obtained with the dnaJ gene (all isolates were correctly identified at the species level). However, some of these isolates achieved a low similarity level (<97%) and required a confirmatory test (sequencing of the rpoB gene). The performance of ID 32 STAPH was poor. Regardless of the probability level used (80% or 90%), the commercial system obtained identification rates <40%. Using MALDI-TOF MS and the commercial Bruker database, 67% of isolates were identified correctly with scores ≥2.0 (acceptable species-level identification) but this number increased to 97% after the database was expanded. The definitive identification of Staphylococcus spp. other than S. aureus causing intramammary infections in cattle often requires a combination of different procedures, and the existing databases should be updated.
Introduction
In many countries, Staphylococcus spp. other than S. aureus are among the main causes of intramammary infections (IMI) in cattle.3,21 These microorganisms constitute a very large and diverse group, varying in pathogen–host relationships, pathogenic properties, and antimicrobial resistance.9,27,30 In addition, some species may carry genes for staphylococcal enterotoxins, posing a risk of food poisoning in humans. 6 As a consequence, accurate species identification is of great importance for understanding not only the overall pathogenicity of these microorganisms for the bovine udder but also the epidemiologic and clinical aspects of an infection at the herd- or cow-level.
Based on previous studies, ~30 Staphylococcus spp. other than S. aureus have been isolated from IMI in cattle.9,12,13,34 Staphylococcus chromogenes is often the most prevalent species causing IMI worldwide, followed by S. simulans, S. xylosus, S. haemolyticus, and S. epidermidis. 31 The remaining species have been isolated less frequently, and their occurrence may often depend on some herd-specific factors, such as barn type, milking system, and herd management program.5,7,18,22
A variety of approaches have been used for species-level identification of Staphylococcus spp. other than S. aureus. Identification methods may vary greatly in terms of accuracy, simplicity of use, cost, and availability of equipment. Regardless of the method used, the value of a method is largely dependent on the comprehensiveness of the database underpinning the method. If the database is not updated appropriately, identification of rare or recently described species as well as atypical isolates of common bacteria can be difficult. Owing to the abundance and considerable diversity of Staphylococcus spp. other than S. aureus, the laboratory identification of this group can still pose a problem. Phenotypic methods are often considered to be of low accuracy because these bacteria exhibit great variability in expression of biochemical properties, and isolates having aberrant features can be unrecognized or misidentified. 23 Genetic methods have proved superior to those based on phenotypic properties. 16 In particular, sequence analysis of some genes has become the gold standard for the accurate identification of Staphylococcus spp. other than S. aureus. 34
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is considered a fast and reliable identification method for various groups of pathogenic microorganisms, including Staphylococcus spp. other than S. aureus causing IMI.4,29 This technology combines some of the advantages of phenotypic tests (relatively low cost and labor expenditure for sample processing) and genotype-based methods (high accuracy). The main drawback of MALDI-TOF MS, apart from the high purchase cost of equipment, may be an insufficient number of reference spectra included in the commercial database. 29 The database can, however, be expanded quite easily in the laboratory to meet specific needs of a user.4,17
The capacity of various identification methods depends to a large extent on the composition of the bacterial population causing IMI in a given area. 34 Species distribution as well as phenotypic and genotypic properties of these microorganisms may vary in different countries; therefore, to fully assess the efficacy of a particular method, comprehensive studies carried out on a large number of staphylococcal species are necessary. We compared and report herein the performance of phenotypic identification using ID 32 STAPH (bioMérieux, Marcy-l’Etoile, France), sequencing of the 16S rRNA and dnaJ genes, and MALDI-TOF MS analysis for the identification of Staphylococcus spp. other than S. aureus causing IMI in cows in Poland.
Materials and methods
Bacterial isolates
We studied 131 staphylococcal isolates recovered from the milk of cows with subclinical or clinical mastitis, collected in the period 2013–2016 on 3 unrelated dairy farms in Lower Silesia, Poland. The isolation of bacteria was performed on blood agar (tryptone soya agar [Oxoid, Basingstoke, UK] with 5% defibrinated sheep blood) incubated aerobically for 24 h at 37°C. Isolates were identified tentatively based on colony and cell morphology, Gram staining, and catalase activity. The isolates were then kept frozen (Cryo-billes; bioMérieux) at −80°C.
Identification by molecular methods
Bacterial DNA was isolated as described previously. 12 Briefly, 2 mL of bacterial culture grown in brain–heart infusion broth (Oxoid) were centrifuged for 5 min at 12,000 × g and resuspended in 100 μL of 100 mM Tris-HCl buffer (Sigma-Aldrich, Poznań, Poland), pH 7.4, containing 10 μg of lysostaphin (A&A Biotechnology, Gdańsk, Poland). After 30 min incubation at 37°C, 10 μL of 10% sodium dodecyl sulfate buffer (Sigma-Aldrich) was added, and the sample was incubated for another 30 min. Two hundred μL of 5 M guanidine hydrochloride (Sigma-Aldrich) were then added, and the sample was vortexed and incubated at room temperature for 10 min. The DNA was extracted with phenol-chloroform-isoamyl alcohol (25:24:1; Sigma-Aldrich), precipitated with ethanol, and dissolved in water.
All of the staphylococcal isolates examined were identified using partial sequence analysis of the 16S rRNA and dnaJ genes (encoding the small subunit of the ribosomal RNA and heat-shock protein 40, respectively). For 16S rRNA amplification and sequencing, the primers 16S-27f (5’-AGAGTTTGATCMTGGCTNAG-3’) and 16S-907r (5’-CCGTCAATTCMTTTRAGTTT-3’) were used, 10 giving a PCR product of ~927 bp. Amplification of the dnaJ gene (a fragment of ~920 bp) was carried out using previously described primers and conditions. 25 PCR products were then purified by adding 10 U of exonuclease I and 2 U of shrimp alkaline phosphatase (Thermo Fisher Scientific, Warsaw, Poland) to 5 μL of the reaction mixture. Sequencing of the products was performed on both DNA strands (DYEnamic ET terminator cycle sequencing kit; Amersham Biosciences Europe, Freiburg, Germany). Sequence similarities ≥99% (for the 16S rRNA gene) or ≥97% (for the dnaJ gene) were regarded as decisive for species identification.13,34 A few isolates that still could not be assigned unambiguously to a species based on these criteria were additionally examined by partial sequencing of the rpoB gene as described previously 12 with 94% similarity as the cutoff value. 15
Phenotypic identification by means of the ID 32 STAPH system
The system was used according to the manufacturer’s instruction (bioMérieux). Identification of bacteria was carried out with apiweb v.3.0 identification software (bioMérieux, https://apiweb.biomerieux.com). To determine the acceptable level of identification, 2 thresholds of high probability levels were compared: 80% (as suggested by the apiweb database) and 90% (adopted by other investigators). 23
MALDI-TOF MS
The isolates were analyzed by MALDI-TOF MS (UltrafleXtreme spectrometer; Bruker Daltonics, Bremen, Germany). Preparation of bacterial isolates was performed using protein extraction with ethanol and formic acid as described previously. 17 According to the manufacturer, the following identification scores were used: <1.7 = no reliable identification; 1.7–1.999 = probable identification to the genus level; 2.0–2.299 = secure genus identification, probable species identification; and 2.3–3.0 = highly probable species identification. Scores ≥2.0 were considered as acceptable species-level identification. 24 Each isolate was run at least twice, and isolates that failed to achieve a score ≥2.0 were retested with a maximum of 4 repetitions.
For 7 staphylococcal species with isolates that failed to be identified using the Biotyper v.3.1 software and preexisting Biotyper reference library (containing 6,904 entries), the v.3.1 database was expanded by inducting 9 main spectrum profiles (MSPs) obtained from 9 isolates examined in our study. Two MSPs were created for S. haemolyticus, covering both subpopulations of that species as described in earlier work, 32 and 2 for S. chromogenes, representing isolates from 2 different farms. In the case of 5 species (S. epidermidis, S. microti, S. rostri, S. agnetis, and S. vitulinus), 1 MSP was added for each species. The procedure of obtaining the MSPs was the same as reported previously. 12 All 131 isolates were analyzed again with the expanded Biotyper database.
Results
Identification by molecular methods
Based on the combined molecular identification, the 131 isolates were assigned to 18 species (Table 1). Staphylococcus haemolyticus (33 isolates) was the species isolated most frequently, followed by S. chromogenes (25 isolates), S. epidermidis (22), S. microti (12), and S. rostri (9). The remaining 13 species were represented much less frequently and comprised 30 isolates in total. Partial sequence analysis of the 16S rRNA gene alone allowed for the unequivocal identification of only 56 isolates (42.7%). The performance of this method was best for S. chromogenes, S. epidermidis, S. auricularis, and S. hominis (all isolates belonging to these species were identified correctly). However, 16S rRNA gene sequencing misidentified 13 isolates (with 99.8–100% sequence similarity, 100% query coverage, and E values = 0.0). In particular, 4 S. haemolyticus isolates were incorrectly recognized as either S. hominis or S. epidermidis, and in the case of S. rostri isolates, the best hit in the BLAST program was S. microti. For the remaining 62 isolates, identification by sequence analysis of the 16S rRNA gene gave ambiguous results—the correct species scored highest in BLAST but 2 or 3 different Staphylococcus species shared sequence similarities >99%. For example, 27 of 33 of our S. haemolyticus isolates had 99.5–100% similarity of the 16S rRNA gene with S. petrasii subsp. jettensis, S. hominis, S. epidermidis, or S. devriesei.
Molecular identification of 131 isolates of Staphylococcus spp. other than S. aureus examined in our study.
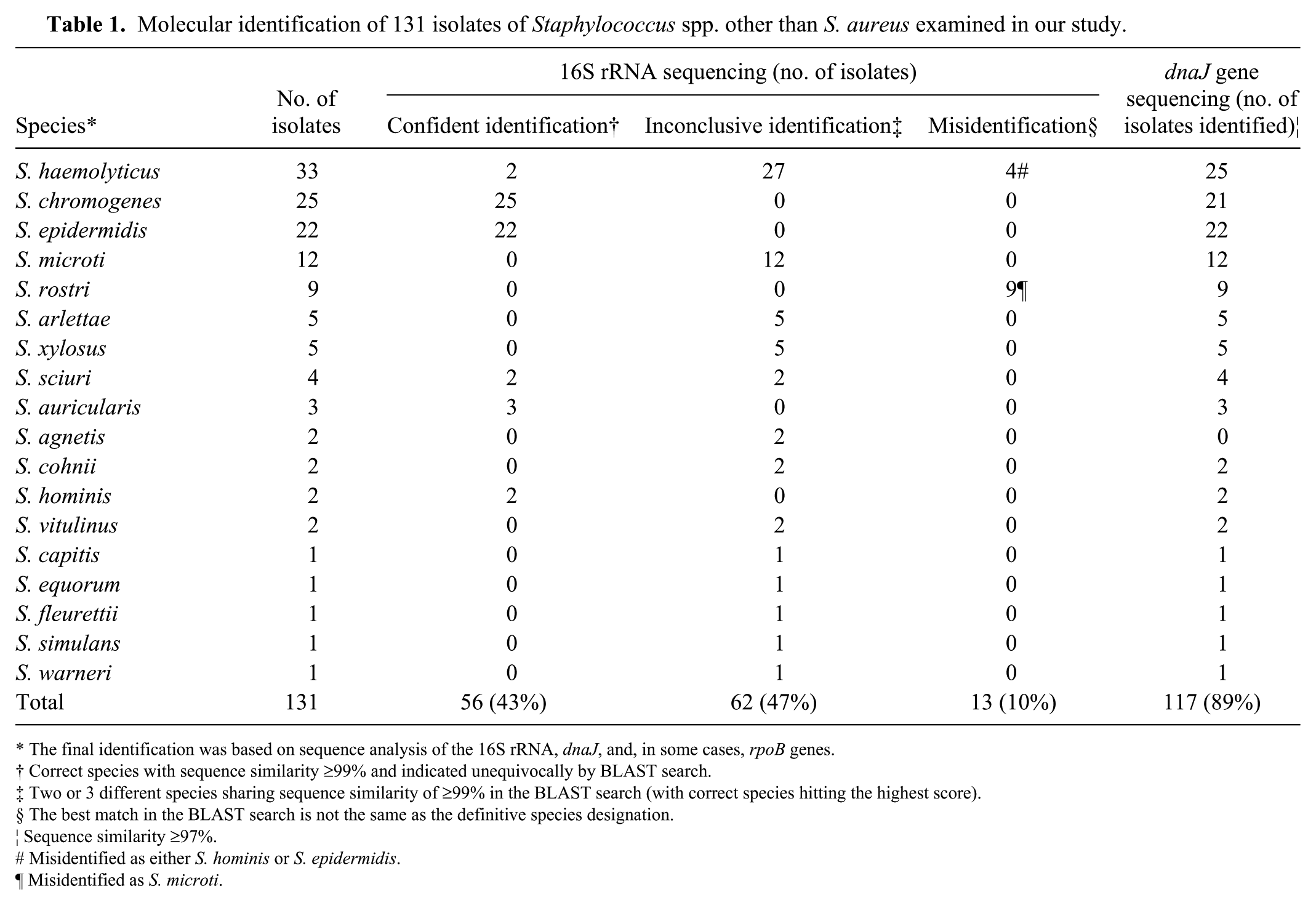
The final identification was based on sequence analysis of the 16S rRNA, dnaJ, and, in some cases, rpoB genes.
Correct species with sequence similarity ≥99% and indicated unequivocally by BLAST search.
Two or 3 different species sharing sequence similarity of ≥99% in the BLAST search (with correct species hitting the highest score).
The best match in the BLAST search is not the same as the definitive species designation.
Sequence similarity ≥97%.
Misidentified as either S. hominis or S. epidermidis.
Misidentified as S. microti.
Sequence analysis of the dnaJ gene proved to be much more efficient for the identification of Staphylococcus spp. other than S. aureus. Although all of the isolates were identified correctly, the stipulated level of similarity ≥97% was achieved for 89% (n = 117) of isolates. Eight S. haemolyticus isolates had sequence similarity of the dnaJ gene of 92.5–92.8%. Similarly, 4 S. chromogenes and 2 S. agnetis isolates displayed 96% and 94%, respectively, of sequence similarity to known sequences of these species. These 14 isolates were examined additionally by sequence analysis of the rpoB gene, achieving sequence similarities of 95.0–99.0%.
Identification by means of ID 32 STAPH
By setting the probability level of acceptable species identification at 80%, the results of the ID 32 STAPH system matched those obtained by the combined molecular identification in 52 of 131 isolates (40%; Table 2). Of the remaining isolates, 63 were unidentified and 16 were misidentified (48% and 12%, respectively). The method was most efficient for S. xylosus, S. cohnii, and S. simulans (all of the isolates were identified correctly). However, a much poorer performance of ID 32 STAPH was observed for the 3 species most commonly found in our study (i.e., S. haemolyticus, S. chromogenes, and S. epidermidis; 30%, 40%, and 68% of isolates, respectively, were identified). The system failed to identify isolates belonging to S. microti, S. rostri, S. agnetis, S. vitulinus, and S. fleurettii (none of these species are included in the apiweb identification software), as well as S. capitis, S. equorum, and S. warneri (all of these species are included in the identification software).
Identification of 131 Staphylococcus spp. other than S. aureus using ID 32 STAPH (bioMérieux).
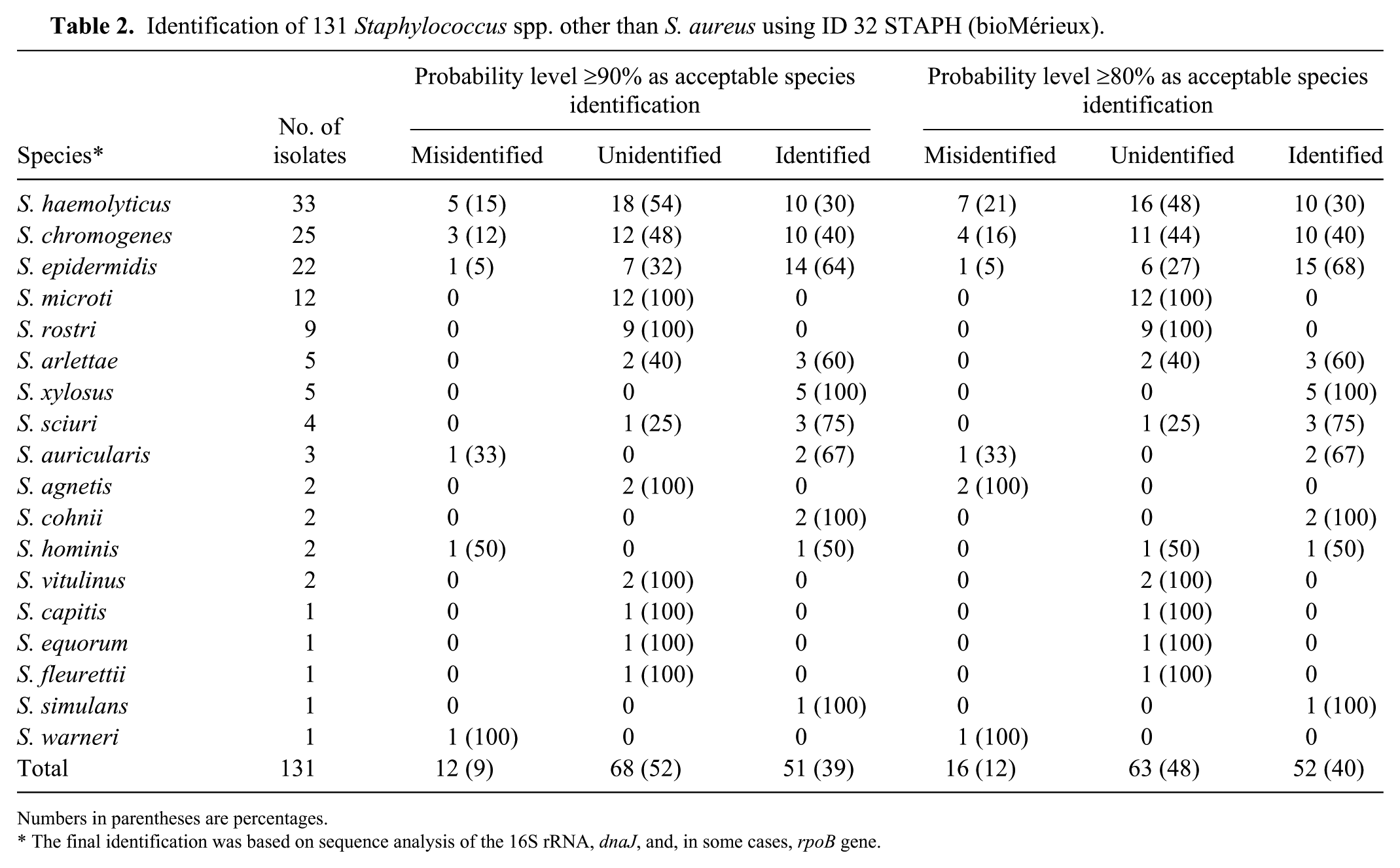
Numbers in parentheses are percentages.
The final identification was based on sequence analysis of the 16S rRNA, dnaJ, and, in some cases, rpoB gene.
Increase of the probability level of an acceptable species identification to ≥90% had only a minimal impact on the number of isolates identified correctly (51 isolates matched the consensus obtained by sequence analysis of the 3 genes analyzed). However, the increased probability level resulted in a lower number of misidentified isolates (Table 2).
MALDI-TOF MS identification
Using the preexisting Biotyper database, 108 of 131 isolates (82%) were identified correctly; however, only 88 (67%) achieved reliable species-level scores (i.e., ≥2.0; Table 3). Twenty-three isolates (18%) belonged to species not included in the preexisting database (S. microti, S. rostri, S. agnetis) and, consequently, could not be identified. All but one of these unidentified isolates achieved scores <1.7 (no reliable identification) or ≥1.7 and <2.0 (identification to the genus level only). In the case of one isolate, the score obtained was within the range of acceptable species identification but the isolate was named incorrectly by the system (an isolate of S. agnetis, scoring 2.08, and thus identified as S. hyicus). The best performance of the system was observed for S. xylosus, S. sciuri, S. auricularis, S. cohnii, and S. hominis (all of these isolates were correctly identified with scores ≥2.0). In the case of the 3 species most frequently isolated (i.e., S. haemolyticus, S. chromogenes, and S. epidermidis), the acceptable identification to the species level was achieved by 79%, 76%, and 86% of the isolates, respectively. The remaining isolates of these 3 species as well as 2 isolates of S. vitulinus, 1 of S. arlettae, and 1 of S. simulans were correctly indicated by MALDI-TOF MS but their scores were 1.7–2.0 (acceptable identification to the genus level only).
Identification of 131 Staphylococcus spp. other than S. aureus by MALDI-TOF MS (Bruker Daltonics).
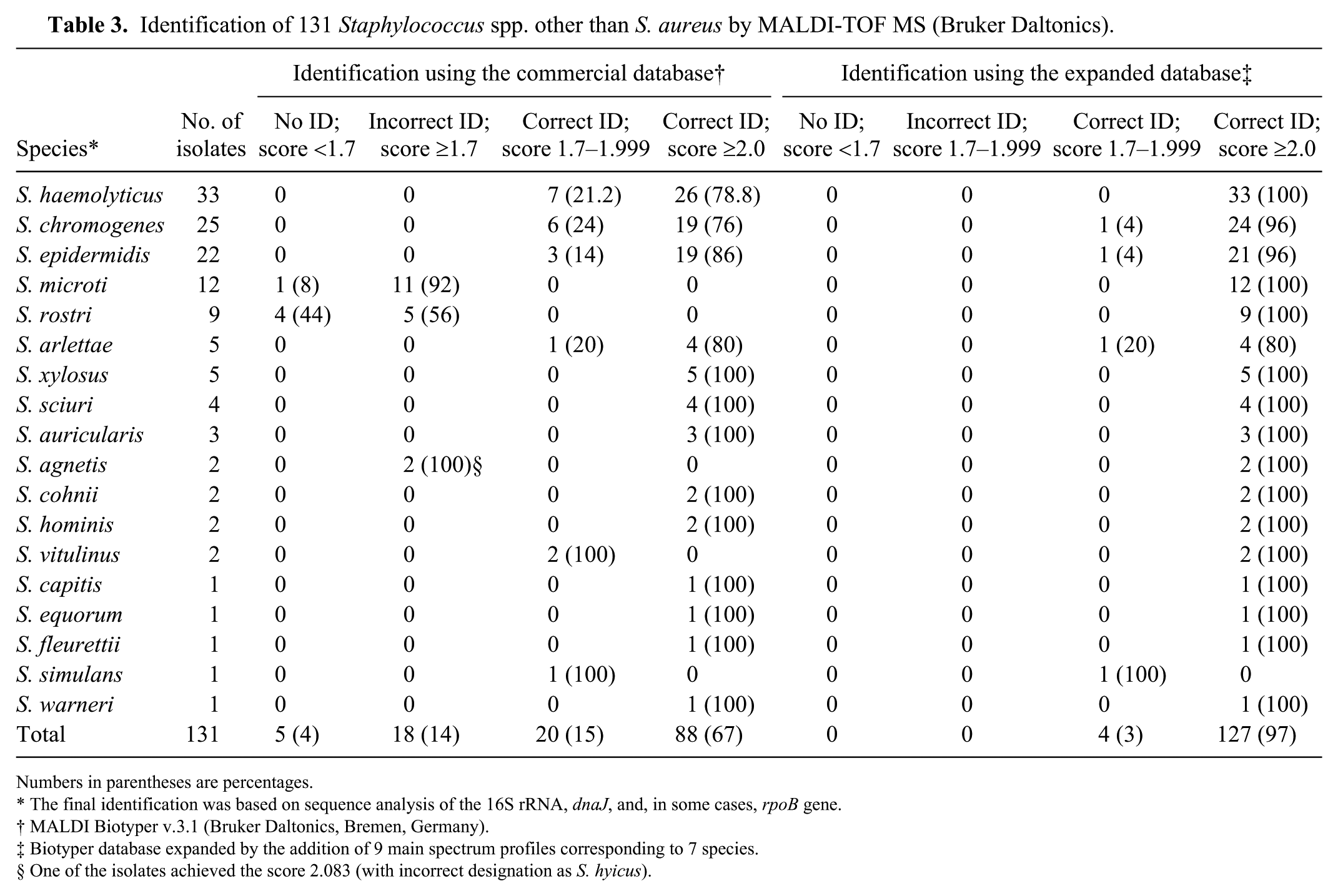
Numbers in parentheses are percentages.
The final identification was based on sequence analysis of the 16S rRNA, dnaJ, and, in some cases, rpoB gene.
MALDI Biotyper v.3.1 (Bruker Daltonics, Bremen, Germany).
Biotyper database expanded by the addition of 9 main spectrum profiles corresponding to 7 species.
One of the isolates achieved the score 2.083 (with incorrect designation as S. hyicus).
After expanding the database with 9 MSPs, the number of isolates identified correctly, that is, having results of MALDI-TOF MS consistent with those of the sequence-based methods and scoring ≥2.0, increased to 127 (96.9%). For 4 isolates (each belonging to a separate species), identification scores remained <2.0.
Discussion
The 3 species detected most frequently in our study (S. haemolyticus, S. chromogenes, and S. epidermidis) belong to the most common Staphylococcus spp. other than S. aureus causing IMI worldwide.4,13,18 However, some of them are also reported to show considerable phenotypic and genetic diversity19,32 and, consequently, may not be sufficiently covered by existing databases. Interestingly, a large group of the isolates examined in our study was assigned to recently described or rarely recognized species, such as S. microti, S. rostri, S. agnetis, and S. vitulinus. Thus, the identification methods that we used were challenged against a relatively broad spectrum of staphylococcal species that may represent different levels of difficulty for the diagnostic laboratory.
In general, genotype-based methods are accepted as the “gold standard” for the species-level identification of Staphylococcus spp. other than S. aureus.4,34 However, molecular techniques may have limitations, including insufficient discriminatory power in the case of closely related species (reported especially for the 16S rRNA gene) or the lack of quality of sequences deposited in GenBank. 11 These drawbacks of the 16S rRNA gene sequencing were also evident in our study. In many cases, the bacteria examined showed a very high level of sequence similarity (often >99.5%) to sequences from 2 or 3 different Staphylococcus species.
The problem of a relatively low discriminatory power of the 16S rRNA gene in the case of some staphylococci has also been reported by other investigators.25,26,33,34 Another study 28 detected a high degree of homology of this gene between S. agnetis and S. hyicus (99.6–99.7%) as well as between S. agnetis and S. chromogenes (99.1%), so that discrimination of these species was practically impossible. This finding is of practical significance considering the fact that S. agnetis, S. chromogenes, and S. hyicus have been isolated (with various frequencies) from cases of IMI in cattle.1,28 A similar high level of interspecies homology of the 16S rRNA gene has been observed for S. capitis and S. caprae 16 and for S. xylosus and S. saprophyticus. 13 Noteworthy, despite these failings, both groups of investigators reported very good performance of partial 16S rRNA sequencing, with accuracy of 98.5% and 93.6%, respectively.13,16 Such disparities between results obtained by various authors may result from differences in the composition of bacterial isolates examined as well as from some differences in methodology (e.g. various lengths of DNA fragments) and in interpretive criteria (e.g., different cutoff levels for species delineation). The overall low performance of the method, detected in our study, can be attributed to a relatively large number of isolates belonging to unusual (S. microti, S. rostri) or more heterogeneous species (e.g., S. haemolyticus).
Given that sequence analysis of selected “reference” genes is considered to be a much more reliable method than 16S rRNA gene sequencing for species identification of Staphylococcus spp. other than S. aureus,26,34 we sequenced the dnaJ gene of all of the isolates that we examined. This was supported by sequencing of the rpoB gene for those isolates that failed to achieve the recommended cutoff values of ≥97% similarity for the dnaJ gene. The dnaJ gene was chosen because it has been shown to be more discriminative than some other conserved genes, 25 permitting, for example, clear discrimination between the 2 closely related species of S. microti and S. rostri. 12 Using dnaJ gene sequencing, all 131 isolates were given their correct species designation (100% accuracy); however, 14 isolates (belonging to S. haemolyticus, S. chromogenes, and S. agnetis) did not fulfill the postulated criterion of ≥97% similarity to known sequences of the corresponding species. Therefore, the identity of these 14 isolates was confirmed by sequence analysis of the rpoB gene (97–99% similarity with reference data).
Although rpoB gene sequencing is often used as the reference method for identification of Staphylococcus spp. other than S. aureus,9,23 some limitations of this approach have been identified. Most importantly, rpoB frequently displays a very high level of intraspecies diversity, which is reflected in the fact that some investigators recommend the cutoff value of the gene to be 94%.15,34 This heterogeneity may not be appropriately covered by sequences available in databases, thus generating difficulties in species identification. For example, our previous study revealed that some strains of S. haemolyticus may be more closely related to S. hominis with regard to the rpoB gene. 32 Moreover, rpoB may also have too little discriminatory power in certain instances, as described in a 2017 study. 1 Thus, owing to the possible drawbacks of genotype-based methods, sequencing of 2 or even more genes may often be necessary before reaching a definitive identification.
In contrast to genotype-based methods, the ID 32 STAPH system proved to be unsatisfactory for the identification of the staphylococci examined in our study. The overall performance of the test was only ~39%, with best results obtained for S. xylosus and S. cohnii (although the low numbers of these isolates preclude more reliable conclusions). Roughly half of our isolates could not be identified by ID 32 STAPH, and this fact may be attributed in large part to 2 drawbacks in the database of the system. First, a large group (one-fifth) of the isolates belonged to staphylococcal species not covered by the identification system. This is an increasing concern because new species continue to be recognized and some of them (e.g., S. fleurettii, S. agnetis, and S. nepalensis) have been repeatedly reported from around the world as causative agents of IMI.1,28,34 Second, problems in identification of several well-recognized IMI-linked species, examined within our study, resulted from phenotypic variations in some key biochemical tests. Relatively poor performance of phenotype-based commercial systems in the identification of Staphylococcus spp. other than S. aureus causing IMI in cattle has also been reported by other investigators. For example, the examination (by means of ID 32 STAPH) of 172 isolates collected in The Netherlands 23 gave very similar results to those obtained in our study. By setting the probability level at ≥90% as acceptable for species identification, these authors were able to identify correctly 41% of isolates, whereas 47% were unidentified and 12% misidentified. 23 Much better results, often >80% accuracy, have been reported for staphylococci of human origin.11,14 Such differences can be attributed to various probability thresholds considered for reliable identification or, more frequently, the composition of staphylococcal species examined. Based on our study, a confidence limit of ≥90% seems to be more appropriate than that of ≥80% for species delineation by ID 32 STAPH, because both levels of probability gave similar numbers of correctly identified isolates, but the misidentification rate was lower when the ≥90% criterion was used.
Our results and the literature data highlight the need for extension of the ID 32 STAPH database to obtain greater coverage of bovine isolates. In veterinary medicine, there is hardly any other type of staphylococcal infection in which so many coagulase-negative or -variable species are involved so frequently, as IMI in cattle. Updating the existing database with some animal-specific species (e.g., S. agnetis, S. vitulinus, S. fleurettii) and incorporating a more diversified collection of common species isolated from milk samples (e.g., S. chromogenes and S. haemolyticus) would ensure that the ID 32 STAPH becomes a more valuable method for the identification of Staphylococcus spp. other than S. aureus causing IMI.
In light of the above problems, MALDI-TOF MS offers a useful alternative for accurate and cost-effective identification of pathogens causing IMI in cattle. To date, there are only a few reports describing the application of MALDI-TOF MS for the identification of bovine-associated Staphylococcus spp. other than S. aureus. Although the results of these prior studies are very promising, the method requires further validation and standardization. 4 In our study, 131 bacterial samples were first compared with the commercial Bruker Biotyper 3.1 database. In this preliminary assessment, 108 isolates (82%) were identified correctly; however, only 88 (67%) gave scores ≥2.0. Interestingly, the performance of MALDI TOF MS was relatively poor for the 2 most frequently isolated species in our study (i.e., S. haemolyticus and S. chromogenes; <80% of these isolates achieved acceptable species-level scores). A similar rate of overall accuracy of the system (62.4%) was reported in a study of 258 coagulase-negative Staphylococcus isolates. 4 Using the cutoff of ≥2.0 as acceptable for species identification and the Biotyper commercial database, this earlier study also detected a high number of S. haemolyticus and S. chromogenes isolates producing low-score (≥1.7 and <2) but accurate MALDI-TOF MS results (80.8% and 21.3% of isolates, respectively). 4 This can be attributed to marked heterogeneity of these species, reported especially for S. haemolyticus,19,32 and an insufficient number of species representatives in the library. 4 In fact, the Biotyper database has been developed mainly for human clinical microbiology and is gradually being expanded with veterinary isolates.2,29 The system has been very efficient for identification of a number of staphylococcal species isolated from human clinical specimens as well as food and plant samples,8,14 with a reported species-level identification rate of 99.3%. Good performance of MALDI-TOF MS (77.4% and 95.4%) was also detected in 2 studies concerning coagulase-negative staphylococci isolated from ruminants.20,29 The main drawbacks of the method reported by these previous authors were an inability to successfully identify S. agnetis 20 or lower performance in the case of S. saprophyticus. 29
An important benefit of MALDI-TOF MS is that the commercial database may be easily extended and updated by the user, and the addition of in-house reference spectra will improve the performance of the system.4,17 We added 9 MSPs created for 7 staphylococcal species, based on the number of isolates of a given species not fulfilling the stipulated criterion of “reliable species identification” (score ≥2.0) and the expected level of species heterogeneity. Re-analysis of 131 isolates with the extended database significantly increased (from 67% to 97%) the total number of isolates achieving the species-level cutoff of ≥2.0.
Apart from the need for a comprehensive reference database, the performance of MALDI-TOF MS may also depend on other factors, such as the method of sample preparation and the criteria for data interpretation. 24 According to the manufacturer’s criteria, the cutoff of ≥2.3 indicates “highly probable species identification” and scores of ≥2.0 result in “probable species identification.” This latter cutoff is commonly accepted as a reliable identification to the species level.4,20,24,29 In fact, the cutoff of ≥2.0 seems to be relatively safe for correct identification of staphylococcal species given that only a few examples of isolates incorrectly identified with scores ≥2.0 may be found in the literature (e.g., S. agnetis has been misidentified as S. hyicus). 20 Indeed, some authors postulate a further reduction of the species cutoff.4,24 In one study, the decrease of the species-level score from 2.0 to 1.9 resulted in a higher rate of species-level identifications. 24 Other investigators suggest that lowering the cut-point to 1.7 would improve identification rates without adversely affecting the accuracy of identification of bovine Staphylococcus spp. other than S. aureus. 4 This is, however, not confirmed by the results of our study. Because some staphylococcal species may not be in the MALDI-TOF MS database, a cutoff reduction below 2.0 greatly increases the risk of misidentification.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.