
Abstract
We investigated the feasibility of an assay based on target-specific primer extension, combined with a suspension array, for the multiplexed detection and typing of a veterinary pathogen in animal samples, using Streptococcus suis as a model pathogen. A procedure was established for simultaneous detection of 6 S. suis targets in pig tonsil samples (i.e., 4 genes associated with serotype 1, 2, 7, or 9, the generic S. suis glutamate dehydrogenase gene [gdh], and the gene encoding the extracellular protein factor [epf]). The procedure was set up as a combination of protocols: DNA isolation from porcine tonsils, a multiplex PCR, a multiplex target-specific primer extension, and finally a suspension array as the readout. The resulting assay was compared with a panel of conventional PCR assays. The proposed multiplex assay can correctly identify the serotype of isolates and is capable of simultaneous detection of multiple targets in porcine tonsillar samples. The assay is not as sensitive as the current conventional PCR assays, but with the correct sampling strategy, the assay can be useful for screening pig herds to establish which S. suis serotypes are circulating in a pig population.
Introduction
PCR is a valuable technique that is widely used in diagnosis and control of animal disease. 8 However, PCR and real-time PCR are limited in the number of assays that can be performed in one simultaneous reaction. For multiplex detection of nucleic acid sequences, parallel real-time PCRs on microfluidic arrays are possible, 17 as are comprehensive syndromic microarrays, 15 but both require costly arrays and equipment. Inexpensive microarrays that can be analyzed with inexpensive equipment also exist,4,11 but these assays depend on a fixed probe configuration. Multiplexing can also be set up as a bead-based suspension array for which the probe configuration is not fixed and the required equipment can be acquired at modest cost. A great advantage of bead-based arrays is that a large number of simultaneous assays are possible (i.e., 50, 80, or 500 depending on the equipment used). For these suspension arrays, several assay types are possible, for example hybridization of amplicons upon exponential amplification, or hybridization of single-stranded products upon linear amplification.6,10 The feasibility of target-specific primer extension (TSPE) combined with a suspension array was demonstrated for multiplexed detection of plant pathogens.30,31 We chose a similar approach to investigate multiplexed detection of veterinary targets in samples of animal origin, using Streptococcus suis as a model pathogen.
S. suis is a significant swine pathogen in nearly all countries with an intensive pig industry, and also has considerable zoonotic potential.21,27,35 Over 30 different capsular serotypes of S. suis have been reported.3,19 Although serotype 2 is the most prevalent serotype worldwide in diseased pigs, 12 serotypes 1, 7, and 9 are also frequently found to be associated with diseased pigs. 38 Good laboratory assays to identify S. suis carrier pigs are required to better understand the epidemiology of the disease and to facilitate effective control measures. Many multiplex PCR assays for the detection and (sero)typing of S. suis in clinical samples have been described, but these often rely on enriched samples22,33,36 or on isolates, as reviewed previously. 12 To date, no complete S. suis detection and typing assay has been reported that can be used on DNA isolated from clinical samples to our knowledge.
We set the detection of 6 targets by their nucleic acid sequences as the objective. The selected targets are 4 serotype-specific genes for S. suis serotypes 1, 2, 7, and 9, one serotype-independent generic S. suis gene encoding glutamate dehydrogenase (gdh), and a virulence marker encoding the extracellular protein factor (epf). For the latter, a design was made that exclusively targets the virulent form that has no inserts. 23 We compared the resulting assay (i.e., a combination of a multiplex PCR, a TSPE, and a suspension array) with a panel of conventional PCRs, and determined the strengths and weaknesses of the proposed assay.
Materials and methods
Strains and culturing
Reference strains of all S. suis serotypes 19 were used, complemented with a serotype 2 field strain (strain 3) 32 that is positive for extracellular protein factor (EF+). Further, 31 non–S. suis bacteria from an in-house collection 36 were used (Actinobacillus pleuropneumoniae, Bacillus subtilis, Bordetella bronchiseptica, Campylobacter coli, Enterococcus faecalis, Erysipelothrix rhusiopathiae, Escherichia coli, Haemophilus parasuis, Klebsiella pneumoniae, Listeria monocytogenes, Micrococcus spp., Pasteurella multocida, Proteus vulgaris, Pseudomonas aeruginosa, Salmonella enterica subsp. enterica serovar Typhimurium, Staphylococcus hyicus, S. bovis, S. dysgalactiae, S. dysgalactiae subsp. equisimilis, S. gordonii, Streptococcus group L, S. milleri III, S. mitis, S. mutans, S. oralis, S. pneumoniae, S. pyogenes, S. sanguinis, S. uberis, S. zooepidemicus, Yersinia enterocolitica). Bacteria were grown overnight at 37°C in air with 5% CO2 on 6% horse blood agar plates (Columbia blood agar base, Oxoid, Landsmeer, The Netherlands) or on heart infusion agar plates containing 5% sheep blood and 0.1% nicotinamide adenine dinucleotide (Biotrading, Mijdrecht, The Netherlands; A. pleuropneumoniae and H. parasuis).
Clinical samples
A farrow-to-finish piggery that had recent problems related to S. suis infections was selected. Thirty 8-wk-old pigs and 10 farrowing sows were sampled by tonsillar brushing using a sterile tooth brush. 28 Immediately after sampling, the tooth brushes were transported on ice to the laboratory in 15 mL of Todd–Hewitt broth (Oxoid) with 0.25% Streptococcus selective supplement SR0126 (Oxoid) and 0.2 mg/mL of crystal violet. The tubes were vortexed 5 times for 30 s, and placed for 1 h in an ultrasonic water bath at room temperature. A 1.5-mL aliquot was stored at −80°C in the presence of 15% glycerol.
DNA preparations
DNA of all strains was isolated using a phenol–chloroform extraction method as described. 9 DNA of S. suis serotypes 1, 2, 7, and 9 reference strains was prepared using the MagNA Pure LC total nucleic acid isolation kit and MagNA Pure LC isolation robot (Roche Diagnostics, Almere, The Netherlands), according to the instructions of the manufacturer. To obtain DNA from the porcine tonsillar tooth brushes, a 200-μL subsample from the stored material was lysed using lysostaphin, lysozyme, and mutanolysin (Sigma-Aldrich, Zwijndrecht, The Netherlands) as described. 7 The DNA isolation was finalized with the same kit and robot as described above.
Multiplex PCR design
Using publicly accessible sequences, consensus sequences were created of the regions targeted by published PCR assays aimed at speciation (gdh), 20 virulence (epf), 37 and serotyping (cps1I, cps2J, cps9H, cps7H).24,25 Using conserved regions within these consensus sequences, PCR primer pairs were designed (AlleleID 7.6, PREMIER Biosoft, Palo Alto, CA). The primers were designed to allow amplification of the 6 targets in 1 multiplex reaction (Table 1). Each primer pair was designed for S. suis–specific sequences, except the gdh primers, which also match the gdh of S. pneumoniae. This was taken into account by designing an S. suis–specific primer for the subsequent TSPE as described below. The epf primer pair may also amplify non-functional epf* variants that contain extra sequences within the target region. 23 This was taken into account when designing the subsequent epf-specific TSPE primer (see below). Primers designed for PCR were obtained as unpurified oligonucleotides (Biolegio, Nijmegen, The Netherlands).
Oligonucleotides for PCR and target-specific primer extension (TSPE) of Streptococcus suis strains.

TSPE primers consist of 2 segments (i.e., an xTAG (5’half) and a complement of the target sequence). The xTAG is complementary to the oligonucleotide on the MagPlex-TAG bead (Luminex). The boundary between the regions is indicated by a hyphen.
Design of primers for TSPE
TSPE primers were designed (PrimerPlex 2.0, PREMIER Biosoft) for amplicon production in multiplex PCR reactions (Table 1). For all TSPE reactions, the primers were designed to hybridize to the sense strand, except for epf (see below). To allow use of commercial paramagnetic beads for capture of single-stranded TSPE products, all TSPE primers were designed with specific sequences at their 5’-end (xTAGs) that are compatible with available beads (see below).
For cps genes, straightforward design of TSPE primers was possible. For gdh, the TSPE primer was designed to explicitly target an S. suis–specific single nucleotide polymorphism (3’-end of the oligonucleotide), which in theory enables differentiation between the 2 species S. suis and S. pneumoniae. This is relevant because the preceding PCR will amplify the selected gdh region of both streptococci (see above). For epf, a TSPE primer was designed for the antisense strand using the following strategy: the primer largely overlaps with the forward PCR primer that runs up to a known insertion point (between nucleotides 2859 and 2860 of epf in S. suis strain D282; accession X71881). 23 However, the TSPE primer is 2 nucleotides longer at the 3’-end and reaches over the insertion point. This strategy allows (partial) hybridization of this primer with amplicons from all epf variants, but allows only full hybridization and primer extension when targeted amplicons are from an epf with no inserts. Primers designed for TSPE were obtained as high-performance liquid chromatography–purified oligonucleotides (Biolegio).
PCR
Conventional PCR assays for serotyping (targeting cps1I, cps2J, cps7H, cps9H) and detection (epf, gdh) were performed essentially as described20,24,25,37 using a hot-start polymerase (HotStarTaq Mastermix, Qiagen, Venlo, The Netherlands) on a GeneAmp PCR System 9700 (in 9600 run mode; Applied Biosystems, Life Technologies Europe, Bleiswijk, The Netherlands). Multiplex PCRs were performed using a polymerase dedicated to multiplexing (Multiplex PCR Plus Kit, Qiagen; Fig. 1) or an ordinary hot-start polymerase (HotStarTaq Mastermix, Qiagen; all other results) that is less well suited for multiplexing, but is compatible with the DNA used. All PCRs were performed in 25-μL reactions on a GeneAmp PCR System 9700 (in 9600 run mode): 5 min 95°C, 30 cycles of 30 s at 95°C, 90 s at 50°C, and 90 s at 72°C, followed by 10 min at 68°C. PCR products were analyzed by agarose gel electrophoresis (4% precast E-gel, Invitrogen, Life Technologies Europe) with a molecular weight marker (O’RangeRuler 20 bp DNA Ladder, Invitrogen, Life Technologies Europe) used in each run.
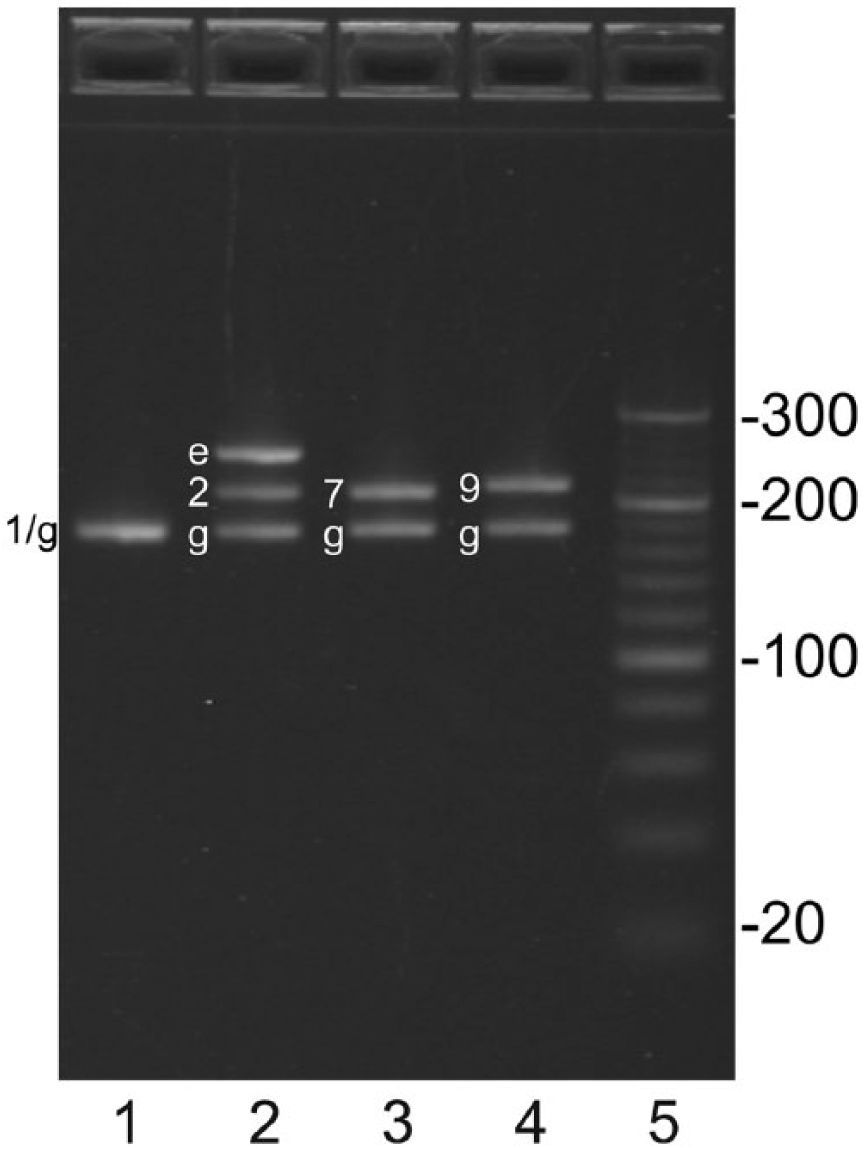
Amplicons generated by multiplex PCR were analyzed on 4% agarose gel. As template, DNA from Streptococcus suis strains of serotype 1, 2 (EF+), 7, and 9 (lanes 1–4) was used. Amplicons generated from genes cps1I, cps2J, cps9H, cps7H, gdh, and epf are labelled 1, 2, 7, 9, g, and e, respectively. For expected sizes of the various amplicons, see Table 1. The sizes of the 20-bp DNA marker (lane 5) are indicated.
TSPE step
After multiplex PCR, excess primers and nucleotides were removed using Sepharose (GE Healthcare, Eindhoven, The Netherlands), as described. 30 The subsequent TSPE was performed as described for allele-specific primer extension. 1 In short, single-stranded biotinylated products were generated using 5 μL of Sepharose-purified PCR product as template, with 6 TSPE oligonucleotides in 1 reaction mixture containing a special polymerase (Platinum Genotype Tsp DNA Polymerase, Invitrogen) and biotinylated nucleotides (Invitrogen). The run parameters were as follows: 2 min 96°C, 30 cycles of 30 s at 94°C, 1 min at 55°C, and 2 min at 74°C.
Hybridization
For detection of TSPE products, a mix was prepared containing paramagnetic beads (MagPlex-TAG beads A027, A033, A056, A064, A072, A073, Luminex, ‘s-Hertogenbosch, The Netherlands) carrying oligonucleotides complementary to the 5’-extensions (xTAGs) of the TSPE primers (Table 1). Hybridization of TSPE products to beads was performed as described. 1 In brief, a 50-μL reaction mixture was prepared in hybridization buffer (0.2 M NaCl, 0.1 M Tris, 0.08% Triton X-100, pH 8.0) containing 5 μL of the TSPE reaction and 1,250 beads of each bead set. In a GeneAmp PCR System 9700 (in 9600 run mode), the mix was denatured (1.5 min at 96°C) and then cooled to allow hybridization of amplicons to beads (37°C for 30 min). The 96-well plates with reaction mixtures were then placed on a magnet (Dynal MPC-96-S, Invitrogen) for 1 min. The supernatant was removed and replaced by 75 µL of streptavidin–phycoerythrin (2 μg/mL; Prozyme, ImmunoSource, Halle-Zoersel, Belgium) in hybridization buffer. The sample was incubated for 15 min at 37°C on a thermal mixer (BioShake iQ shaker, Quantifoil Instruments, Jena, Germany) at 600 rpm.
Analyses of beads were performed at 37°C (Luminex 200 with xPONENT 3.1 software, Luminex) using default settings. For each sample, fluorescence of at least 100 beads per bead set was measured (median fluorescence intensity [MFI]). Where required, cutoffs were calculated for each bead set as the average of the expected negative samples plus 5 times the standard deviation.
Experiments
Evaluation of the multiplex PCR and TSPE was performed using strains of serotype 1, 2 (EF+), 7, and 9. To verify the designed multiplex PCRs, amplicons were generated using a polymerase and buffer combination dedicated to multiplexing, and analyzed by agarose gel electrophoresis. To verify the TSPE designs, amplicons were generated with the designed multiplex PCR using an ordinary hot-start polymerase, and subsequently used in a TSPE reaction in triplicate. The resulting TSPE products were then analyzed using the suspension array.
Specificity of the suspension array (i.e., the combined procedure of multiplex PCR, TSPE, and hybridization to paramagnetic beads) was evaluated with multiple S. suis strains and non–S. suis isolates in comparison with conventional PCR assays. These and all other (see below) multiplex PCRs were performed using the ordinary polymerase.
Analytical sensitivities of the suspension array were compared with that of the conventional PCRs using 10-fold serial dilutions of 4 S. suis strains. These were tested for the 4 serotype-related cps genes. Prior to purification and TSPE, amplicons from the multiplex PCR were also analyzed on agarose gels to check the presence of amplicons as input for TSPE.
Evaluation of the suspension array with field samples was done with 40 tonsillar specimens from pigs originating from a herd with recent S. suis infections. Results of the conventional PCRs were used to define samples positive or negative for a particular target, which in turn was used to calculate cutoffs for each bead set of the suspension array (except for gdh) and to determine the diagnostic sensitivities and specificities.
Results
Evaluation of the multiplex PCR and TSPE
Gel analysis of amplicons from the multiplex PCR showed that amplicons of expected sizes were generated (Fig. 1; Table 1) with one exception. The gdh and cps1I amplicons of the serotype 1 strain are of similar size and were not distinguishable as separate products. Analysis of TSPE products showed that, for each target, the suspension array acquired signals that were in concordance with the genes expected to be present in the test strains (Fig. 2; i.e., for all strains a signal was observed on gdh beads, whereas for each strain the correct cps-specific signal was acquired. As expected, only the serotype 2 (EF+) strain gave a signal for the epf beads).
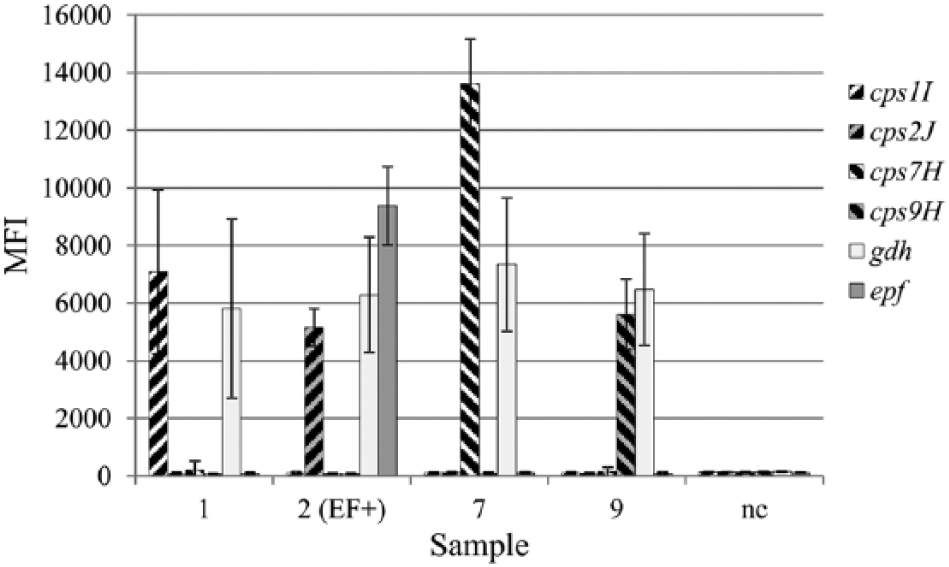
Amplicons generated by multiplex PCR from Streptococcus suis strains of serotype 1, 2 (EF+), 7, and 9 were subjected to target-specific primer extension and subsequently analyzed with the suspension array, using a mix of 6 bead types, each carrying specific probes. For each sample, the results for the 6 bead types are indicated; water was used as negative control (nc). The target-specific primer extension and subsequent hybridizations were performed in triplicate; fluorescent signals are given as median fluorescence intensity (MFI) with standard deviations from the 3 experiments.
Specificity of the suspension array
Evaluation of the suspension array with multiple S. suis strains showed results for cps genes and epf that matched the results obtained by PCR analysis (Supplementary Table 1), and were in concordance with the expected genetic makeup of the strains. For gdh, the suspension array returned substantial signals for the majority of strains, bar serotypes 13, 16, and 17. Further, no significant gdh signals were obtained for strains that in retrospect are not S. suis (the reference strains for serotypes 20, 22, 26, 32–34). In the conventional PCR analysis for gdh, amplicons were generated with all DNA preparations, except for S. suis serotype 13.
When the suspension array was evaluated with non–S. suis isolates (Supplementary Table 2), on all 6 bead sets the acquired signals remained <300 MFI (i.e., below the calculated cutoffs), except for S. pneumoniae, which resulted in a low signal of 341 MFI on the gdh beads. As expected, for 2 positive controls (S. suis serotype 2 EF+ and EF*), the acquired signals for cps2J and gdh were above the cutoffs (calculated within the set), as was the signal for epf for S. suis serotype 2 (EF+), all in concordance with the genetic makeup of these strains.
Analytical sensitivities of the suspension array
For serotypes 1 and 2, the occurrence of positive gdh signals in the suspension array matched the presence of amplicons prior to TSPE and hybridization (Supplementary Table 3). For serotypes 7 and 9, the suspension array was more sensitive (one step in the serial dilution) than the multiplex PCRs. Some cross-reactions were observed, such as (low) signals of serotype 9 on the cps7H bead, and of serotype 2 on the cps1I bead. PCRs for cps and epf genes were more sensitive than the multiplex PCR and suspension array.
Evaluation of the suspension array
Of 40 tested field samples, 10 were positive in the conventional cps1I PCR; these samples were always positive in 2 or more conventional cps PCRs (Supplementary Table 4). More than half (23 of 40) of the samples were PCR-positive for cps2J, and, except for sample 11, PCR-positive for epf (22 of 23). By conventional PCR, 23 samples were positive for cps7H; all of these samples were also PCR-positive for cps9H, and most samples were also PCR-positive for cps1I and/or cps2J. In fact, most field samples (38 of 40) were PCR-positive for cps9H. Although all 40 field samples were PCR-positive for at least 1 cps gene, the conventional gdh PCR was negative for 5 of 40 samples.
All signals in the suspension array that are designated as positive, were also positive in the corresponding conventional PCR, but for a large number of results from conventional PCR assays, the positive result could not be reproduced by the bead-based assay (Supplementary Table 4). In comparison with conventional PCR, the new assay resulted in low diagnostic sensitivities for cps genes (0.30, 0.42, 0.61, 0.61 for cps1I, cps2J, cps7H, cps9H, respectively) at high diagnostic specificities of 1.00, given that there were no false-positives in the suspension array (Table 2). For epf, diagnostic sensitivity of the suspension array was 0.91, also at a diagnostic specificity of 1.00. Diagnostic sensitivity and specificity for gdh were not available; a cutoff for the gdh bead in the suspension array could not be calculated because true S. suis–negative field samples (i.e., PCR-negative for gdh and all cps genes) were not present in the set tested. Nevertheless, the observed high MFI values on the gdh beads suggested that S. suis was present in all samples, concordant with the observation that all samples were positive for gdh and/or at least one cps gene by conventional PCR.
Diagnostic sensitivity (DSe) and specificity (DSp) of the suspension array.
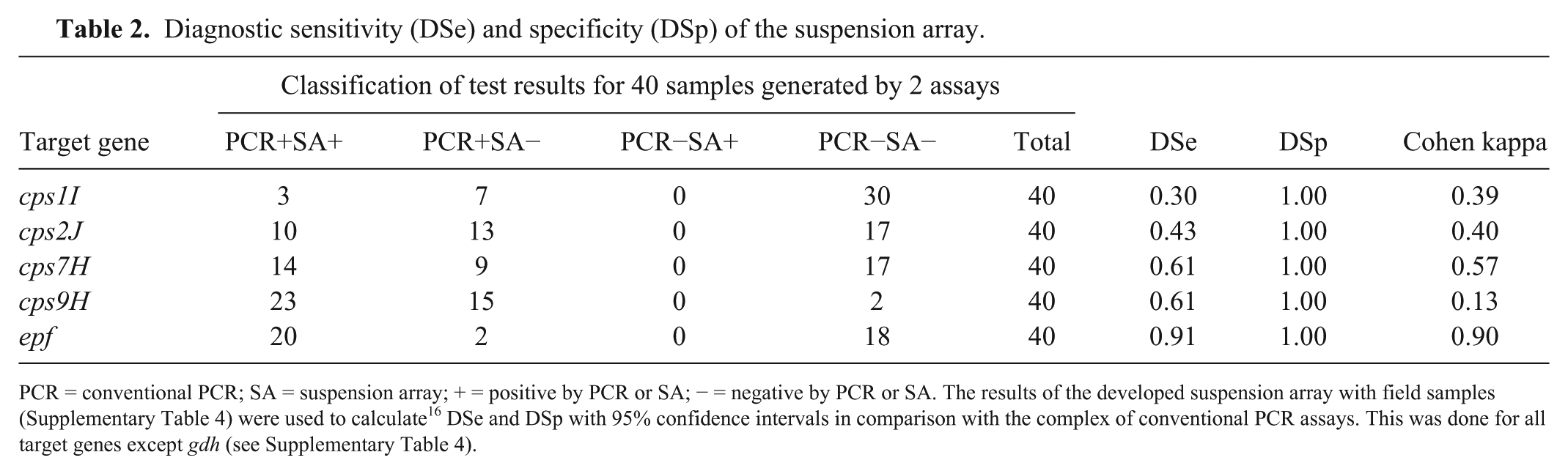
PCR = conventional PCR; SA = suspension array; + = positive by PCR or SA; − = negative by PCR or SA. The results of the developed suspension array with field samples (Supplementary Table 4) were used to calculate 16 DSe and DSp with 95% confidence intervals in comparison with the complex of conventional PCR assays. This was done for all target genes except gdh (see Supplementary Table 4).
Discussion
Using DNA from S. suis reference strains, we confirmed that the proposed assay is sufficiently specific for the cps genes, despite the occurrence of some minor cross-reactions. This shows that the new assay can be used to serotype isolates. The design for epf was not extensively tested with all possible epf variant strains. 23 However, the results show that only a serotype 2 EF+ strain returns a signal, whereas a serotype 2 EF* strain, containing an insert in epf, correctly remains negative in the suspension array.
Concerning detection of gdh, for several of the reference strains used, there is doubt if these strains are S. suis.13,18,29 This situation is confirmed by our observation that the disputed S. suis serotypes 20, 22, 26, 32, 33, and 34 reference strains are indeed gdh negative by the suspension array. Three true S. suis strains were missed by the suspension array (the reference strains for serotypes 13, 16, and 17), of which the serotype 13 reference strain was also negative by conventional gdh PCR. At the time of design, all available S. suis gdh sequences matched with the TSPE primer. However, a newly added serotype 16 strain 34 indeed has a gdh sequence that differs at the TSPE primer binding site, showing that the current design for the gdh primers may need revision. The difference in results between the conventional and multiplex PCRs shows that correct speciation using gdh depends on the region chosen as target, indicating that unambiguous speciation of S. suis is difficult. As an example, a species-specific PCR based on an alternative gene such as fbpS recognizes S. suis serotypes 1–34, 26 which is in conflict with previous reports on speciation, where for particular serotypes it is disputed if they are S. suis.13,18,29
The analytical sensitivity of the conventional PCRs is greater than that of the new multiplex assay. In fact, the conventional PCR is more sensitive than the multiplex PCR part of the assay, even though the same quality of DNA and the same polymerase was used. True multiplexing, with multiple targets present in a sample, was possible with a dedicated PCR mix that is optimized for multiplexing, and resulted in discrete products and similar amounts of amplicons for each targeted gene sequence (Fig. 1). However, the routine procedure used at our laboratory to isolate DNA from tonsillar brush samples, which is based on a commercial DNA isolation kit and the corresponding robot, yielded DNA that was not compatible with a PCR kit dedicated to multiplexing. As a compromise, for all experiments with the suspension array, the amplification was performed with a different, but comparable, PCR kit from the same manufacturer. As this kit is not optimized for multiplexing, this resulted in less efficient and biased DNA amplification, and, as a consequence, reduced sensitivity of the developed multiplex assay. Thus, regarding analytical sensitivity, there is room for improvement by changing the DNA isolation to a method compatible with the use of a polymerase dedicated to multiplexing.
Results with the complex of conventional PCRs showed that, based on the presence of cps signals, all 40 tonsil samples tested were S. suis positive, and that most samples harbored a mix of S. suis strains (Supplementary Table 4). There was a discrepancy with the conventional gdh PCR given that only 35 of 40 samples were gdh positive, another indication that S. suis identification using the gdh gene is challenging. For the gdh component of the new assay, no diagnostic sensitivities or specificities were calculated because there were no S. suis–negative samples. The results of the serotyping by conventional PCRs could not be reproduced by the new assay (Table 2). Nonetheless, in our sample survey, the test correctly identified the presence of S. suis serotypes 1, 2, 7, and 9 in a herd from a farm with recent problems related to S. suis infections with serotypes 1, 2, and 9. The assay also indicated that strains with serotype 7 were present, a situation not known before our study.
Use of imperfect assays for classification of herds is possible 5 and has been applied for identification of Mycobacterium avium–positive herds with an ELISA that has a low sensitivity. 14 Therefore, even though the sensitivities of the suspension array for S. suis are not very high for each separate target gene (Table 2), the assay may be used to screen herds for circulating S. suis serotypes. Setting cutoffs for field application is an issue that still needs to be resolved; ideally, a large set of samples would be used, and analyzed with a reference method that can be used to set fixed thresholds.
A 2-step suspension array for serotyping S. suis isolates has been described, 2 wherein the initial multiplex PCR primers are used that carry the sequence required for hybridization, thereby forfeiting the need for a TSPE reaction. This approach will significantly increase the assay speed, but it remains unknown if sensitivity is sufficient for field samples. Our 3-step suspension array is not sensitive enough yet, but a better combination of DNA isolation procedure and multiplex polymerase may improve that, as discussed above. It is expected that with the developed 3-step suspension array, analysis of field samples will require less hands-on time and will be faster than analysis by conventional PCRs and gel electrophoresis, especially when larger numbers are being analyzed.
Footnotes
Acknowledgements
We thank Kitty Maassen (National Institute for Public Health and the Environment, RIVM), Jelle de Jong (Wageningen Bioveterinary Research), and Richard van Hoof (RIKILT) for helpful discussions on assay development; Astrid de Greeff (Wageningen Bioveterinary Research) for non–S. suis DNA isolates; PREMIER Biosoft for access to PrimerPlex 2.0; and Hilde Smith (Wageningen Bioveterinary Research) for critical reading of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was partly funded by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement FP7-228394 (NADIR).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.