
Abstract
We compared a qualitative in-clinic (IC)-PCR for the detection of Mycoplasma haemofelis DNA with the results of a commercial qualitative laboratory-based, conventional (c)PCR. In order to determine the specificity of both tests, Bartonella spp. samples were included. Forty-three previously tested blood samples with known PCR results for hemoplasmas and Bartonella spp. were selected. The samples were split between 2 laboratories. At the first laboratory, DNA was purified and run on 2 cPCR assays for the detection of hemoplasmas and Bartonella spp. At the second laboratory, DNA was purified using 2 purification protocols and both run in the IC-PCR assay. The cPCR results confirmed that 18 samples were positive for M. haemofelis, 5 for ‘Candidatus M. haemominutum’, 8 for Bartonella henselae, 2 for Bartonella clarridgeiae, and 10 were negative for both genera. No mixed infections were observed. The IC-PCR assay for the detection of M. haemofelis had a sensitivity of 94.4% and specificity of 96%, when using the same DNA purification method as the first laboratory. Using the second purification method, the sensitivity of the IC-PCR assay was 77.8% and specificity was 96%. Bartonella species were not detected by the IC-PCR M. haemofelis assay. The IC-PCR assay decreased the amount of time to final result compared to a cPCR assay.
Cats are known to be infected by at least 3 hemoplasmas: Mycoplasma haemofelis, ‘Candidatus Mycoplasma haemominutum’, and ‘Candidatus Mycoplasma turicensis’. Subclinical infection and hemolytic anemia are the most commonly recognized syndromes in infected cats. Generally, the most severe clinical disease syndromes are attributed to M. haemofelis, with the other 2 agents typically not inducing hemolytic anemia unless concurrent disease is present. 7
Traditionally, feline hemoplasma infections were detected by cytologic evaluation of blood smears to detect the pleomorphic bacteria on the erythrocytes of cats.6–8 However, cytology has been found to be a problematic diagnostic tool, as false-positives and false-negatives are common, with improper staining resulting in artifacts (false-positives) and varying parasitemia levels occurring in clinically ill animals (false-negatives).6–8 For a number of years, PCR assays have been employed and can be designed to amplify and differentiate the 3 hemoplasmas.6,9 Because of specialized equipment needs and quality control issues, PCR assays for veterinary use have generally only been offered in research or diagnostic laboratories.
We compared the results of a new qualitative detection system designed to amplify M. haemofelis DNA as an in-clinic (IC)-PCR assay (PCRun feline mycoplasma molecular detection kit, Biogal Galed Laboratories, Kibbutz Galed, Israel) to the results of a conventional (c)PCR assay offered in a commercial diagnostic laboratory in the United States (Center for Companion Animal Studies, Colorado State University, Fort Collins, CO). We also determined whether the IC-PCR assay amplifies the DNA of Bartonella spp. that are commonly present in the blood of cats. 3
The electronic records system in the Center for Companion Animal Studies was searched for feline samples that had been assayed for DNA of the hemoplasmas and Bartonella spp. using previously published cPCR assays.5,6 The 43 samples selected were based on referred sample sets (positive or negative for the target organisms) and sample availability. The samples had been stored at −80°C in temperature-monitored freezers until used in our study.
On the day the assays were performed, the blood samples in EDTA were removed from −80°C and allowed to thaw completely at room temperature (~22°C). Once thawed, the samples were briefly vortexed followed by a brief centrifuge step, and then aseptically pipetted into 2 tubes: 1 containing 200 μL and 1 containing 120 μL. The 120-μL aliquots were placed at 4°C and shipped the following day (within 24 h) on ice packs to Biogal Galed Laboratories for testing with the IC-PCR assay.
Total DNA was purified from the 200 μL blood aliquot using a commercial kit (Blood mini kit, Qiagen, Valencia, CA), according to the manufacturer’s instructions, with a final elution of 200 μL. All 43 DNA samples were processed in a single PCR run, employing a previously described cPCR assay for hemoplasmas that amplifies DNA of M. haemofelis and ‘Candidatus M. haemominutum’ and a previously described cPCR assay for amplification of Bartonella spp. DNA.5,6 The feline DNA samples were run along with appropriate positive (dual-positive DNA confirmed with sequencing as M. haemofelis/M. haemominutum 6 and B. clarridgeiae/B. henselae 5 ) and negative (no DNA template added) controls. Sterile phosphate buffer was employed in lieu of blood for negative controls during the DNA extraction process. The cPCR assays for both hemoplasma DNA or Bartonella spp. DNA were performed as previously described5,6 with the following changes to both assays: a final PCR reaction volume of 25 μL was used in both assays, and final PCR amplicon visualization was done using a fluorescent dye (6X EZ-Vision one dye, Amersco, Solon, OH) as per the manufacturer’s specifications (Gel Doc EZ gel documentation system, Bio-Rad, Hercules, CA).
Positive amplicons from both PCR assays were gel extracted (QIAquick gel extraction kit, Qiagen) and submitted for sequencing with appropriate primers for each PCR assay 5,6 at a commercial laboratory (Proteomics and Metabolomics Facility, Colorado State University, Fort Collins, CO) to confirm the species identification. All sequenced products were analyzed for homology by comparison to sequence data available in NCBI GenBank using the BLAST database (http://www.ncbi.nlm.nih.gov/).
Upon arrival at the second laboratory and without delay, DNA was purified from the blood samples by 2 DNA purification protocols. The first protocol (DNeasy blood & tissue kit, Qiagen), a filter-based column method, utilized 50 µL of blood for extraction purposes and 200 µL of molecular-grade water for elution, while following the manufacturer’s instructions. This protocol included a heat denaturation step (10 min) and several washes, and required ~35 min to complete. In the second protocol (PCRun sample prep, Biogal), 50 µL of blood were added to a closed outlet plug with a passive 100-µm pore frit filter (MobiTec, Goettingen, Germany) that contained 50 µL of the kit extraction buffer composed of chelators, buffers, and mild detergents (PCRun extraction buffer, Biogal). The column was incubated at 95°C in a basic heat block for 5 min, and DNA was eluted by centrifugation (1,500 × g, 1 min) into 180 µL of the PCRun kit dilution buffer composed of a buffer mix compatible for use with the IC-PCR. During the centrifugation process, 20 µL of crude DNA was released into a collection vial that contained 180 µL of dilution buffer to give a final volume of 200 µL of diluted DNA. The extract was employed “as is” for the amplification reactions. The second extraction protocol required a shorter heat denaturation step (5 min) and direct elution of the DNA without any wash steps. The entire extraction process was completed in <10 min.
The DNA samples from each of the 2 purification methods were evaluated in the IC-PCR assay (PCRun feline mycoplasma molecular detection kit, Biogal) following the manufacturer’s instructions. When using the DNA from the first purification method (DNeasy blood & tissue kit), the PCR reaction pellets were dissolved in 15 µL of PCRun reaction buffer followed by 5 µL of extracted DNA. When employing DNA extracted by the second purification method (PCRun sample prep), the PCR reaction pellets were dissolved with 20 µL of DNA extracted into the PCRun dilution buffer. Each of the suspect samples was assayed once, and a single positive and negative control was added to each of the 14 test runs. Reactions were carried out at a constant temperature of 60°C for 1 h in a small assay reader (weight 1.3 kg; 94 × 206 × 165 mm; PCRun reader, Biogal). The reader is based on a combined platform of a heating unit and a bio-illuminator that can contain up to 16 samples per run. Results can be observed in real-time as a histogram plot.
The infection status of the 43 samples was defined by the hemoplasma and Bartonella spp. cPCR results (Table 1). The analysis results generated from DNA sequencing of the positive cPCR amplicons were employed to define the species. The sensitivity and specificity of the IC-PCR assay was determined by comparison to the results of the cPCR assay for both of the DNA purification protocols utilized at the second laboratory. Based on the cPCR assays and the sequencing results, 33 samples carried a single species of DNA: 18 samples with M. haemofelis, 8 samples with B. henselae, 5 samples with ‘Candidatus M. haemominutum’, and 2 samples with B. clarridgeiae. Ten samples were negative for DNA of both genera. The 10 Bartonella spp.–positive samples were not amplified by the IC-PCR assay or the cPCR assay for hemoplasmas.
Comparison of the results of the in-clinic PCR assay for Mycoplasma haemofelis and conventional PCR assays for hemoplasmas and Bartonella spp. for 43 samples.
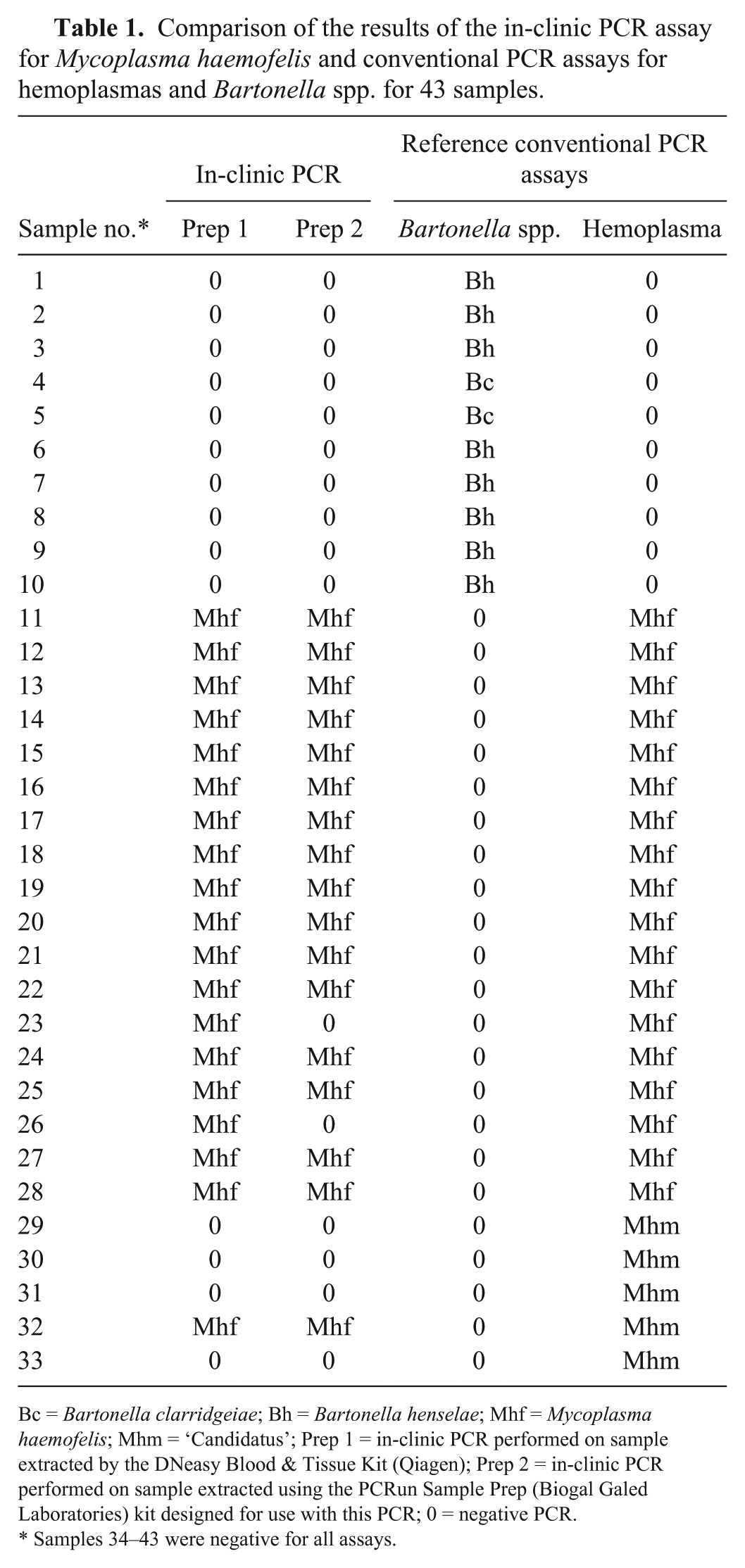
Bc = Bartonella clarridgeiae; Bh = Bartonella henselae; Mhf = Mycoplasma haemofelis; Mhm = ‘Candidatus’; Prep 1 = in-clinic PCR performed on sample extracted by the DNeasy Blood & Tissue Kit (Qiagen); Prep 2 = in-clinic PCR performed on sample extracted using the PCRun Sample Prep (Biogal Galed Laboratories) kit designed for use with this PCR; 0 = negative PCR.
Samples 34–43 were negative for all assays.
Using the first DNA purification protocol (DNeasy blood & tissue kit), the IC-PCR assay for amplification of M. haemofelis DNA had a sensitivity of 94.4% and specificity of 96.4%. Using the second protocol (PCRun sample prep), the IC-PCR assay for amplification of M. haemofelis DNA had a sensitivity of 77.8% and specificity of 96.4%. The difference in sensitivity between the cPCR and IC-PCR may be explained by the difference in DNA extraction blood volumes utilized in the 3 methods. Both extraction protocols performed at the second laboratory for the IC-PCR utilized only 50 µL of blood, whereas the cPCR extraction at the first laboratory used 200 µL of blood. All 3 DNA extraction methods had a final elution of 200 µL. Depending on the efficiency of all of the extraction methods, there is a possibility that a 4-fold increased concentration of DNA was added to each cPCR reaction in comparison to the IC-PCR assay.
The proprietary primers for the isothermal amplification reactions included in the IC-PCR assay for amplification of M. haemofelis DNA target a 230-bp conserved region derived from the 16S ribosomal (r)DNA gene. Based on the results of our study, the IC-PCR assay does not amplify DNA of B. henselae or B. clarridgeiae, which can have an even higher prevalence rate than M. haemofelis in regions endemic for Ctenocephalides felis, the known vector.1–3 This high specificity is important because, although Bartonella spp. infections are common and involve erythrocytes, these organisms are not known to cause hemolytic anemia. 4
The specificity of the IC-PCR assay for amplification of M. haemofelis DNA when compared to the cPCR assay results was high at 96% regardless of the DNA purification protocol. Sample 32 (Table 1) defined as containing only ‘Candidatus M. haemominutum’ DNA in the cPCR assay was positive for M. haemofelis DNA in the IC-PCR assay, leading to a specificity of <100%. The same sample was tested using a commercial PCR kit (genesig standard kit for M. haemofelis and M. haemocanis, Primerdesign, Chandler’s Ford, UK) that targets a 97-bp section of the M. haemofelis 16S rDNA gene. The product from that assay was subjected to sequencing, which revealed 100% homology to M. haemofelis and 75% homology to M. haemobos. An explanation for this discordant result is that the sample actually contained DNA of both organisms, but in the cPCR assay, the reaction was preferentially driven toward ‘Candidatus M. haemominutum’ giving false-negative results for M. haemofelis. If this hypothesis is correct, then the IC-PCR assay would have a specificity of 100% when compared to the cPCR used in our study. Other hemoplasma PCR assays could have been performed in an attempt to further evaluate this hypothesis, but were not performed because additional assays were not in the initial experimental design and therefore not performed in parallel with the other assays, making any result potentially inaccurate for comparison. It would be beneficial to test more samples to further evaluate the potential for discordant results.
The sensitivity (94.4%) of the IC-PCR assay for amplification of M. haemofelis DNA was greater using the first DNA purification protocol (DNeasy blood & tissue kit ) than the second DNA purification protocol (77.8%; PCRun sample prep). The IC-PCR assay can use either extraction protocol; however, the second protocol is less expensive and considerably faster.
Footnotes
Declaration of conflicting interests
S Maurice and T Yaaran are employees of Biogal, Galed Labs. Acs Ltd. Dr. Maurice was masked to the results of the conventional M. haemofelis PCR assay and genetic sequencing until after the in-clinic PCR assays were completed. The remaining authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was financed by Biogal, Galed Labs. Acs Ltd.