
Abstract
Pooled testing of samples is a common laboratory practice to increase efficiency and reduce expenses. We investigated the efficacy of 2 published SYBR Green real-time PCR assays when used to detect the haplosporidian parasite Bonamia ostreae in pooled samples of infected oyster tissue. Each PCR targets a different gene within the B. ostreae genome: the actin 1 gene or the 18S rRNA gene. Tissue homogenates (150 mg) of the New Zealand flat oyster Ostrea chilensis were spiked with ~1.5 × 103 purified B. ostreae cells to create experimental pools of 3, 5, and 10. Ten positive replicates of each pool size were assayed twice with each PCR and at 2 different amounts of DNA template. The PCR targeting the actin 1 gene was unable to reproducibly detect B. ostreae in any pool size. Conversely, the 18S rRNA gene PCR could reproducibly detect B. ostreae in pools of up to 5. Using a general linear model, there was a significant difference in the number of pools that correctly detected B. ostreae between each PCR (p < 0.01) and each pool size (p < 0.01). It is likely that the single copy actin 1 gene is more likely to be diluted and not detected by pooling than the multi-copy 18S rRNA gene. Our study highlights that validation data are necessary for pooled sample testing because detection efficacy may not be comparable to individual sample testing.
The haplosporidian parasite Bonamia ostreae was first reported in New Zealand from the New Zealand flat oyster Ostrea chilensis in 2015. 11 B. ostreae is listed by the World Organization for Animal Health (OIE) as a pathogen that has caused significant disease within farmed and wild flat oyster populations throughout Europe and North America.5,7 After a significant range extension, it is now present within New Zealand—only reported from the Marlborough Sounds 11 —and a Controlled Area Notice has been implemented within this area by the Ministry for Primary Industries (MPI) to prevent the spread of B. ostreae to other New Zealand flat oyster beds. In addition to this, the MPI initiated a national B. ostreae surveillance plan to monitor oyster beds to ascertain the geographic spread of this important parasite.
A nation-wide sampling effort generates a high number of laboratory samples for testing, which is expensive. To reduce this cost as well as the effort of testing, we investigated pooled testing for B. ostreae. A theoretical limitation of pooling is a loss of test sensitivity as a result of either inhibition of the test reaction or dilution of the analyte. 1 However, some cases have shown that pooling samples can actually increase sensitivity, especially at the population level.15,20
Studies have been reported of pooled testing for pathogens affecting livestock, such as Mycobacterium avium subsp. paratuberculosis (MAP) 20 and bovine viral diarrhea virus. 15 The interpretation of pooled test results should be made using sensitivity (Se) and specificity (Sp) values that have been calculated or estimated for that particular testing procedure and for the pool size being used. 21 Laboratories often implement pooling without prior validation. For example, a 2009 study 13 used pooled testing during surveillance for infectious salmon anemia virus, but the authors noted that the effect of pooling on test performance was uncertain. Similarly, the effects of pooled testing for B. ostreae have not been investigated, and the OIE does not recommend pooling until such data become available.
PCR is accepted as the most sensitive and rapid detection method for B. ostreae. 6 There are 2 B. ostreae species-specific SYBR real-time PCR assays currently available,17,18 as well as other non–species-specific Bonamia real-time PCRs.2,12 Both B. ostreae–specific assays were published with extensive accompanying validation data for individual sample testing; however, the effects of pooling on efficacy were not explored. We tested for B. ostreae in an early detection surveillance context (i.e., maximum sensitivity of the assay is desirable when the quantity of the target is expected to be low, with no clinical signs or lesions to support detection). Standard testing procedure is followed in evaluating the efficacy of 2 species-specific SYBR Green real-time PCR assays to detect B. ostreae in pools of 3, 5, and 10 individuals.
Bonamia ostreae–positive O. chilensis previously collected from the Marlborough Sounds 11 and Bonamia-negative O. chilensis collected from the Chatham Islands during the MPI B. ostreae targeted surveillance were used in our study. Whole oysters were not available, so only gill and mantle tissue was used to purify B. ostreae from infected O. chilensis. Following parasite purification10,14 and 0.4% trypan blue (Gibco, Thermo Fisher Scientific, Waltham, MA) staining, 5 × 103/500 µL of Bonamia cells were quantified using a Malassez hemocytometer under a light microscope (Olympus BX51, Olympus New Zealand, Auckland, New Zealand). Using 150 µL of this suspension, DNA was extracted (QIAamp DNA mini kit, Qiagen, Valencia, CA), quantified (Qubit 3.0, Thermo Fisher Scientific), and confirmed with an 18S rRNA gene real-time PCR (Applied Biosystems TaqMan exogenous internal positive control reagents, Life Technologies, Carlsbad, CA). Purified cells were identified as B. ostreae by sequencing a 208-bp amplicon of the B. ostreae 18S rRNA gene. 17 No Bonamia exitiosa was detected.
After B. ostreae purification, we homogenized 10 Bonamia-negative O. chilensis oysters. The tissue was homogenized to enable greater accuracy when producing pools. Gill and mantle tissue (~150 mg) was placed in 600 µL of phosphate-buffered saline (Thermo Fisher Scientific) and homogenized (MagNA lyser, Roche Diagnostics, Sandhofer, Mannheim, Germany) twice for 30 s at 5,000 × g. Once homogenized, the first homogenate was selected as the “B. ostreae–positive” oyster and spiked with ~1.5 × 103 B. ostreae cells. This is well below the calculated 50% lethal dose of B. ostreae 9 and B. exitiosa 4 at 8 × 104 and 11 × 104, respectively, and reflects a low target quantity expected when testing for early detection of B. ostreae. The remaining 9 oyster homogenates were not spiked and were used as “B. ostreae–negative” oysters.
Pool sizes of 3, 5, and 10 were created to test positive for B. ostreae. All pools were created using aliquots from the “B. ostreae–positive” homogenate and, depending on pool size, either 2, 4, or 9 “B. ostreae–negative” homogenates. Following standard procedure of extracting nucleic acid from ~100 µL of homogenate for testing, pools were prepared to a similar volume (i.e., for pools of 3: 33 µL of the positive oyster was pooled with 66 µL of 2 negative oysters; for pools of 5: 20 µL of the positive oyster was pooled with 80 µL of 4 negative oysters; and for pools of 10: 10 µL of the positive oyster was pooled with 90 µL of 9 negative oysters). Ten positive replicates were created for each pool. The B. ostreae–positive homogenate was vortexed between creating each pool replicate to evenly distribute B. ostreae cells throughout the homogenate and ensure accurate distribution of B. ostreae. Once prepared, nucleic acid was extracted (QIAamp DNA Mini kit, Qiagen) from each pool along with the B. ostreae–positive homogenate (i.e., the positive oyster) and each homogenized oyster used to create that pool (i.e., the negative oyster). All extracted nucleic acid was quantified (Qubit 3.0, Thermo Fisher Scientific) and confirmed (Applied Biosystems TaqMan exogenous internal positive control reagents, Life Technologies) as above.
Each pool was assayed with 2 real-time B. ostreae SYBR PCR assays (Ssoadvanced universal SYBR Green mix, Bio-Rad Laboratories, Hercules, CA). The 2 published SYBR assays target different regions of the B. ostreae genome: the actin 1 gene 18 and the 18S rRNA gene. 17 Local thermocycling conditions for the actin 1 gene assay was 98°C for 2 min, followed by 40 cycles of 98°C for 5 s, 60°C for 10 s, and a melt step of 60–95°C for 5 s. The 18S rRNA gene assay had similar conditions except for an annealing temperature of 55°C and a melt step of 55–95°C.
The 10 replicates of each pool size were tested twice with each PCR in 2 separate runs, except for pools of 3 assayed with the actin 1 gene PCR, which was run 4 times and was unsuccessful in obtaining reproducible results. All PCRs used 2 different DNA template amounts of 2 µL and 5 µL. Threshold cycle (Ct) value and melting temperature (Tm) were recorded for all pools tested. A positive result was determined by the Tm being within the predetermined range of 85 ± 0.5°C for the actin 1 gene assay and 83 ± 0.5°C for the 18S rRNA gene assay. For all PCR runs, molecular-grade water (Sigma-Aldrich, St. Louis, MO) was used as PCR template for the negative control, and B. ostreae–infected tissue at 10 ng/µL was used as template for the positive control. The number of Bonamia cells within the genomic DNA extracted from the B. ostreae–infected tissue is uncertain; however, the lower Ct values of the positive control suggest that there are more Bonamia cells than the 5 × 103/500 µL purified in our study.
The individual oysters (i.e., the positive spiked oyster and negative oysters) all produced expected results and were removed from the dataset for statistical analyses so only the pools were analyzed. All statistical analyses were performed in R. 16 A general linear model (GLM) and Bayesian general linear model (BGLM) were used to test for the main effects on the detection of B. ostreae. Under the GLM, all main effects were significant: there were differences in the proportions of positive and negative across the levels of all of the factors, including pool size (p < 0.01); DNA template amount (p < 0.01); and PCR assay (p < 0.01). This is corroborated by the GLM estimate values (data not shown)—where a negative logit value represents a reduction in the probability of detecting B. ostreae, and vice versa—and the marginal plots that visualize the change in probability of detecting B. ostreae in response to a change in a factor averaged across all of the levels (Fig. 1). No interaction between the main effects was observed under the BGLM.
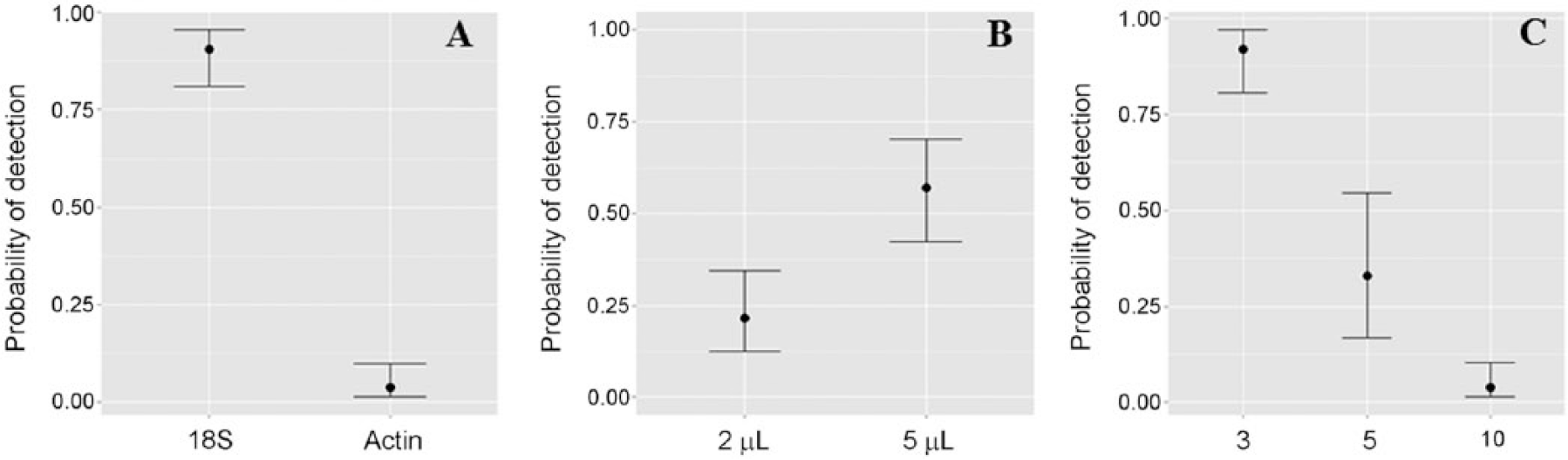
Marginal plots of the probability of detecting Bonamia ostreae as a function of:
Using only data from the 5-µL template, because it detected significantly more B. ostreae than the 2-µL template across both assays (Fig. 1B), the actin 1 gene assay was only able to detect up to 80% of B. ostreae in pools of 3, 30% in pools of 5, and 0% in pools of 10. Conversely, the 18S rRNA gene assay detected 100% of B. ostreae in pools of 3 and 5, and 60% in pools of 10 (Table 1). The significant difference between the performances of the 2 assays likely explains the high variance shown for pools of 5 in Figure 1C because the plots average the results across both PCR tests.
Threshold cycle (Ct), melting temperature (Tm), and PCR results for 10 replicates across 2 runs for each pool size tested with the 18S ribosomal RNA gene assay (only the 5-μL template is shown).*
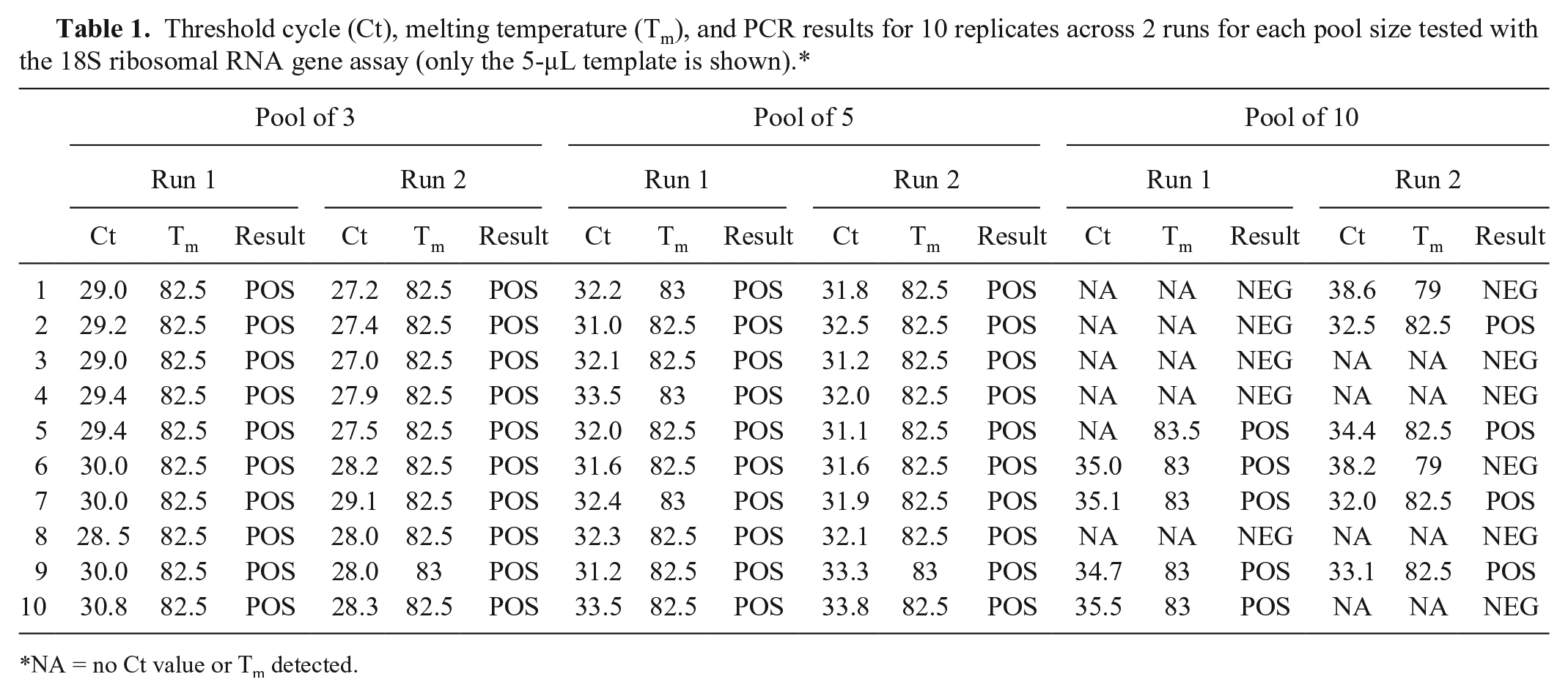
NA = no Ct value or Tm detected.
Excluding the actin 1 gene assay because it did not perform well enough for inclusion in the following analysis, the epidemiologic calculators (http://epitools.ausvet.com.au) estimate that, with the calculated 18S rRNA gene assay test sensitivity, 50 pools of 3 would be required to detect B. ostreae at 2% prevalence with 95% confidence. Testing at the same level, 30 pools of 5 and 26 pools of 10 would be required. For comparison, the MPI B. ostreae targeted surveillance required 150 individual oysters to be collected and tested from each location to test for B. ostreae at the same level. Therefore, all 3 pool sizes used in our study would reduce the number of tests required to test for B. ostreae at this level. Despite the 18S rRNA gene assay having a lower estimated test sensitivity for pools of 10 individuals compared to pools of 3 or 5 individuals, testing in pools of 10 required the fewest number of pools. However, taking into consideration the higher number of individuals initially needed to be sampled for pools of 10 individuals (i.e., 260), it may be more viable to test in pools of 5 individuals because only 4 more pools are required given the higher test sensitivity of the 18S rRNA gene assay at this pool size. Changing from a sample size of 150 individuals to 30 pools of 5 individuals would significantly reduce the amount of laboratory reagents used and laboratory effort expended.
The actin 1 gene assay was unable to reproducibly detect B. ostreae in any experimental pools. The intra-assay coefficient of variation for both assays is low: 2.73% for the 18S rRNA gene and 2.96% for the actin 1 gene assay, therefore, these results are most likely to be caused by dilution of the target gene during pooling, where the likelihood of detecting B. ostreae decreases as the pool size increases. The actin 1 gene is most likely a single copy locus within the B. ostreae genome and, therefore, more vulnerable to the effects of dilution than the multi-copied 18S rRNA gene. 19 Given the inability of the actin 1 gene assay to detect B. ostreae in pools of 3, subsequent troubleshooting led to implementing the 18S rRNA gene assay and, thus, comparing the 2 PCRs.
Using genomic B. ostreae DNA as template, the 18S rRNA gene assay had a reproducible detection limit 2 orders of magnitude lower at 1 × 10-3 than the actin 1 gene assay, 1 × 10-1, which is likely to be influenced by the frequency of the target. Similar results have been observed in a study 19 comparing 3 MAP real-time PCR assays; the multi-copy (20–30 copies) IS900 PCR had a lower limit of detection than the triple-copied ISMAV2-PCR or the single-copied F57 PCR. One disadvantage of a high copy number genetic target is that it can be prone to false-positives. 3 In such cases, screening is recommended with the more sensitive assay, and confirming identification with a lower copied, more specific assay. 20 Both of the real-time assays used in our study were published with extensive validation data,17,18 and it is not surprising that all DNA sequences were specifically B. ostreae.
The actin 1 gene assay performs as the published validation data suggests 18 when testing individual samples, and it has been used extensively with success during the MPI B. ostreae targeted surveillance (H Lane, pers. obs., 2016). This is corroborated by the reproducible detection of B. ostreae within the individual spiked oyster. An upshot of the differences in sensitivity between these PCR assays is that target genes need to be considered carefully during assay design. Furthermore, and more importantly, the performance of an assay during individual testing does not necessarily equate to its performance during pooled testing. It has been surmised 1 that the Se and Sp values of an individual test are unlikely to be the same as those of a pooled test (PSe and PSp, respectively). The authors 1 explain that PSe will likely be lower than Se, especially when the true prevalence within a population is low and pool sizes are large. Moreover, the PSp should exceed that of Sp because dilution should make it less likely to have a false-positive pooled test than a false-positive individual test.
Our findings indicate that pooling 5 individuals is a viable option for detecting B. ostreae using the 18S rRNA gene assay. It is important to note that until PSe and PSp is calculated for pooled testing of B. ostreae, pooling should be applied cautiously. Test validation for pooled testing can be complex, time-consuming, and expensive, but a 2014 published experimental approach 8 enables PSe and PSp to be calculated in accordance with OIE guidelines. In light of this, work is ongoing to build upon our study’s data and generate reliability parameters of PSe and PSp for pooled testing of B. ostreae, which will likely be influential in validating pooled testing for other important aquatic pathogens in the future.
Footnotes
Acknowledgements
We thank the Animal Health Laboratory, Wallaceville, Ministry for Primary Industries, who provided laboratory resources during this work, which was carried out as part of a PhD project at the University of Otago, within Prof. Robert Poulin’s laboratory group. We acknowledge Tim Jowett for his significant contribution to the statistical analyses. We thank the reviewers for their helpful comments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding for this work came from the Zoology Department, University of Otago through a PhD student budget, as well as laboratory consumables supplied by the Animal Health Laboratory, Ministry for Primary Industries.