
Abstract
We report a rapid and reliable method for the detection of Toxoplasma gondii in meat and animal tissues based on real-time polymerase chain reaction (PCR). Samples were collected from cattle, small ruminants, horses, and pigs raised or imported into Sicily, Italy. All DNA preparations were assayed by real-time PCR tests targeted to a 98-bp long fragment in the AF 529-bp repeat element and to the B1 gene using specific primers. Diagnostic sensitivity (100%), diagnostic specificity (100%), limit of detection (0.01 pg), efficiency (92–109%), and precision (mean coefficient of variation = 0.60%), repeatability (100%), reproducibility (100%), and robustness were evaluated using 240 DNA extracted samples (120 positives and 120 negative as per the OIE nested PCR method) from different matrices. Positive results were confirmed by the repetition of both real-time and nested PCR assays. Our study demonstrates the viability of a reliable, rapid, and specific real-time PCR on a large scale to monitor contamination with Toxoplasma cysts in meat and animal specimens. This validated method can be used for postmortem detection in domestic and wild animals and for food safety purposes.
Toxoplasma gondii is an obligate, intracellular protozoan parasite with a wide distribution in Europe.6,7 All warm-blooded animals are susceptible to the infection, and it is estimated that ~80% of the human population in the European Union has been exposed to the parasite. 8 However, given the lack of uniformity in surveillance systems throughout EU member countries, it is very difficult to compare prevalence data. Infection with T. gondii occurs via 3 main routes: congenitally by transmission of the tachyzoite form of the parasite during primary infection of the mother; by ingestion of food or water contaminated with oocysts shed with cat feces; or by ingestion of raw or undercooked meat containing the bradyzoite form in tissue cysts. 3 Seroepidemiologic studies provide evidence of T. gondii infection in meat-producing and wild animals. 4 Infection of meat-producing animals can be correlated with a higher risk of contamination of foodstuffs such as sausages, salami, and cured meats thus resulting in potential risk of human infection following consumption of contaminated foods. Based on the epidemiologic evidence that the main route for human infection is via tissue cysts found in raw or undercooked meat, our goal was to validate a method to detect T. gondii contamination of animal specimens and common meat products by detection of a specific DNA target. Several polymerase chain reaction (PCR)–based techniques have been developed for the detection of toxoplasmosis. Among these techniques, nested PCR is sensitive and reliable but it is time consuming and not practical for high-throughput screening.
The advent of innovative qualitative and quantitative real-time PCR techniques has proven useful in various applications, including pathogen detection and gene expression investigations. Real-time PCR takes advantage of the 5′ nuclease activity of Taq DNA polymerase to cleave a nonextendible fluorescence-labeled hybridization probe during the extension phase of PCR. The fluorescence of the intact probe is quenched by a second fluorescent dye. 1 Nuclease cleavage of the hybridization probe during the PCR releases the effect of quenching, resulting in an increase of fluorescence proportional to the amount of PCR product, and can be monitored by a thermocycler including a fluorescence detector. 9
We report the development and validation of a real-time PCR method for the detection of T. gondii in various biological samples. Analyses were performed in the laboratories of Ce.Tox (Italian National Reference Center for Toxoplasmosis) and of the Molecular Biology Department, both part of the IZS of Sicily (Istituto Zooprofilattico Sperimentale della Sicilia) accredited by Accredia (Italian National Accreditation Body).
Muscle, heart, eyeball, and brain samples were collected from slaughtered cattle, small ruminants, swine, and horses, or in the course of postmortem examination. Thirty positive and 30 negative samples of each tissue were analyzed, for a total of 240 assays. Toxoplasma gondii genomic DNA a was used as reference material for a positive control in each test. We considered true positives and true negatives those samples that tested positive and negative to the nested PCR method recommended by the World Organization for Animal Health (OIE). 12 Five grams of each sample were cut into small pieces and homogenized in 0.1× TE (Tris–EDTA) b buffer using a stomacher c for 120 s at maximum speed. Subsequently, 4 mL of the filtrate were transferred into 5-mL tubes, d and 100 μL of homogenate were then transferred into plates for automatic extraction e using an extraction kit. f DNA extracts were then quantified by spectrophotometric measurement, stored at +4°C, and subsequently tested by both the developed real-time PCR and the confirmatory nested PCR assay. To check DNA quality and to be sure that the extracted DNA was detectable, a housekeeping gene (mitochondrial cytochrome b gene) was amplified using primers common to all mammalian species. A 289-bp long DNA fragment was obtained using the primers g CytB1 (5′- CCAATGATATGAAAAACCATCGTT-3′) and CytB2 (5′-GCCCCTCAGAATGATATTTGTCCTC-3′), 10 with the following conditions: initial denaturation for 5 min at 95°C, 40 cycles of 95°C for 1 min, annealing at 55°C for 1 min, and polymerization at 72°C for 1 min. The reaction mix was prepared in a 50-µL final volume containing 1× master mix, e 10 mM dNTP (2′-deoxynucleoside 5′-triphosphates) mix, 20 pmol of each primer, and 100 pg of DNA target. Finally, the amplicons were detected by a capillary electrophoresis system.
For detection of T. gondii, a 529-bp DNA repeat element (AF) dispersed in the parasite genome 5 was chosen as target. Although we determined the limit of detection (LOD) value, the real-time PCR test was developed as a qualitative method including 2 replicates for positive and negative controls, respectively. The assay was performed in a 20-μL final volume containing 4 μL of master mix 5 X, e added to nuclease-free water (9.6 μL), e 10 pmol of each primer AF1 (CACAGAAGGGACAGAAGT) and primer AF2 (TCGCCTTCATCTACAGTC), g 8 pmol of the labeled TaqMan probe AF 529 (6FAM-CTCTCCTCCAAGACGGCTGG-BHQ), and 100 pg of DNA template. The amplification protocol was performed h as follows: 10 min incubation at 95°C for the hot start, and 40 cycles of a 3-step amplification: denaturation 95°C for 10 s, annealing 55°C for 20 s, and extension at 72°C for 30 s. Each batch of PCR assays included T. gondii DNA a as positive control template and water as negative control template.
Validation parameters were chosen in compliance with OIE 13 and Codex Alimentarius 2 guidelines on performance criteria and validation of methods for the detection of nucleic acids in biological specimens and foods. The following validation parameters were evaluated: diagnostic sensitivity (DSe) and specificity (DSp), LOD, efficiency and precision, repeatability (intra- and interassay), reproducibility, and robustness.
To calculate sensitivity, specificity, and repeatability, 60 samples (30 positive and 30 negative) for each examined biological specimen, equally divided among all species, were tested. DNA extracted from these samples was stored and treated as true-positive and true-negative samples, respectively, to validate the real-time PCR test and to compare the analytical results with those obtained directly on the negative and positive (T. gondii DNA diluted at 100 pg/μL) controls.
A nested PCR targeted to the B1 gene was performed in order to confirm and compare test results. Template DNA was added to a final volume of 50 μL of PCR mixture consisting of 5 μL of 10× PCR buffer, 2 μL of 10 mM deoxy-nucleoside triphosphate, 0.5 μL of Taq DNA polymerase (5 U/μL), i and 20 pmol of each forward primer: B1 up (5′-CGTCCGTCGTAATATCAG-3′) and B1 down (5′-GACTTCATGGGACGATATG-3′). g The following amplification protocol was performed i : the mixture was denatured at 94°C for 10 min, followed by 30 PCR cycles at 94°C for 1 min, 60°C for 15 s, and 72°C for 45 s. One microliter of the resulting PCR product was reamplified under identical conditions in a reaction mixture identical in composition to that of the first-round PCR, except that the following primers g were used: B1 nested forward (5′-GGGAATGAAAGAGACGCTAATGTG-3′) and B1 nested reverse (5′-CTTTTCGCCAGCAGAGGG-3′). The second PCR product was analyzed by electrophoresis j on 2% agarose gel. j
To determine the method detection range, LOD was detected on 6 points of serial decimal dilution between 1,000 pg/μL and 0.01 pg/μL of T. gondii DNA a in genomic extracts isolated from the negative samples, to reveal the impact of potential classes of inhibitors on the LOD values (Fig. 1). PCR amplification efficiency was calculated from 6-fold serial dilutions of the standard T. gondii DNA analyzed in triplicate by plotting the threshold cycle values against the logarithm of the DNA concentration (Fig. 2).e,g,11 Assay efficiency was determined as the slope of the regression line according to the equation: E = 10[−1/slope] − 1. PCR precision was determined by assessing the mean coefficient of variation (CV) over each dilution point of the standard DNA tested in triplicate in a single experiment (Table 1).
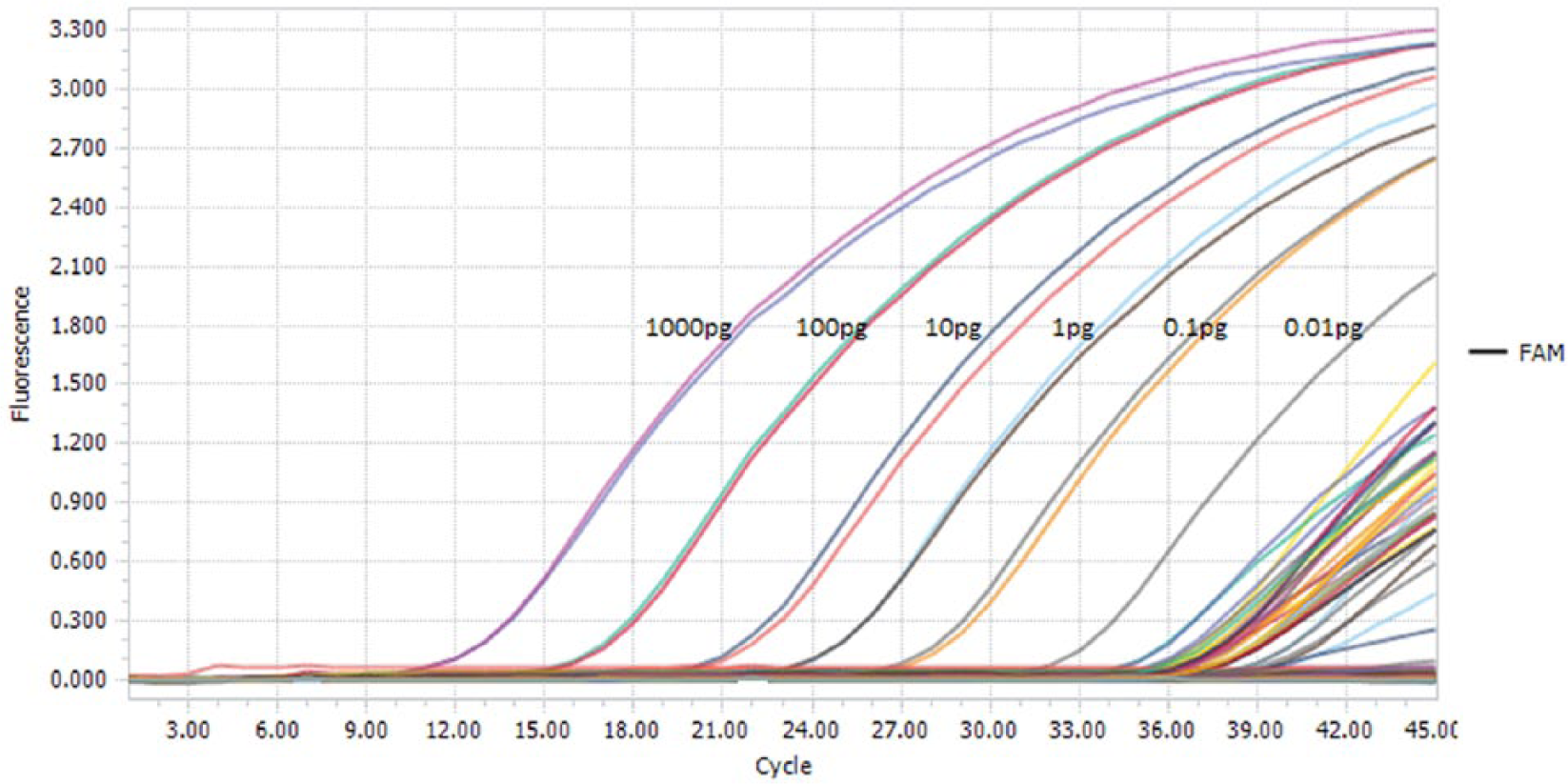
Plot displaying the serial dilutions of Toxoplasma gondii (DNA a ) in a range of 1,000 pg/μL to 0.01 pg/μL and samples with DNA concentration below the lower limit of detection of the standard DNA at 0.01 pg/μL.
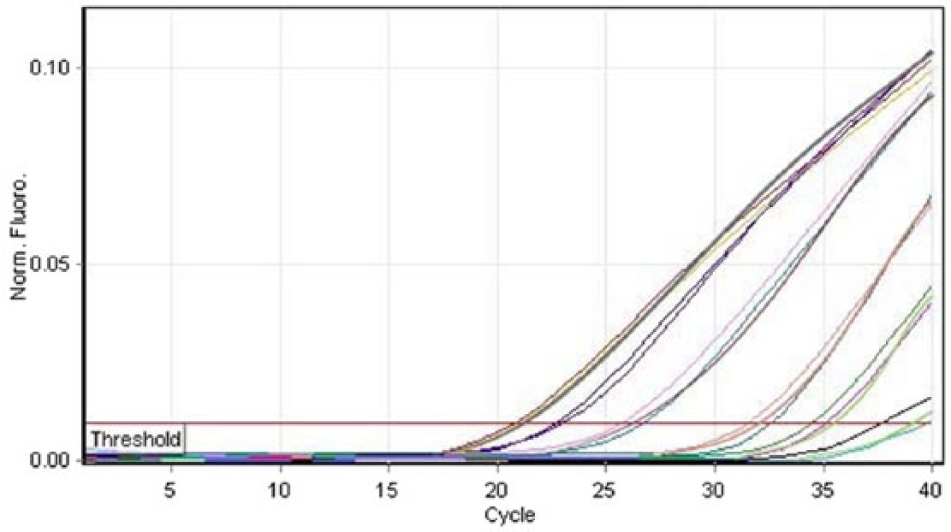
Real-time polymerase chain reaction (PCR) plot showing the analysis of Toxoplasma gondii standard DNA dilutions in triplicate.
Threshold cycle (Ct) values for each decimal dilution of the standard DNA tested in triplicate.*
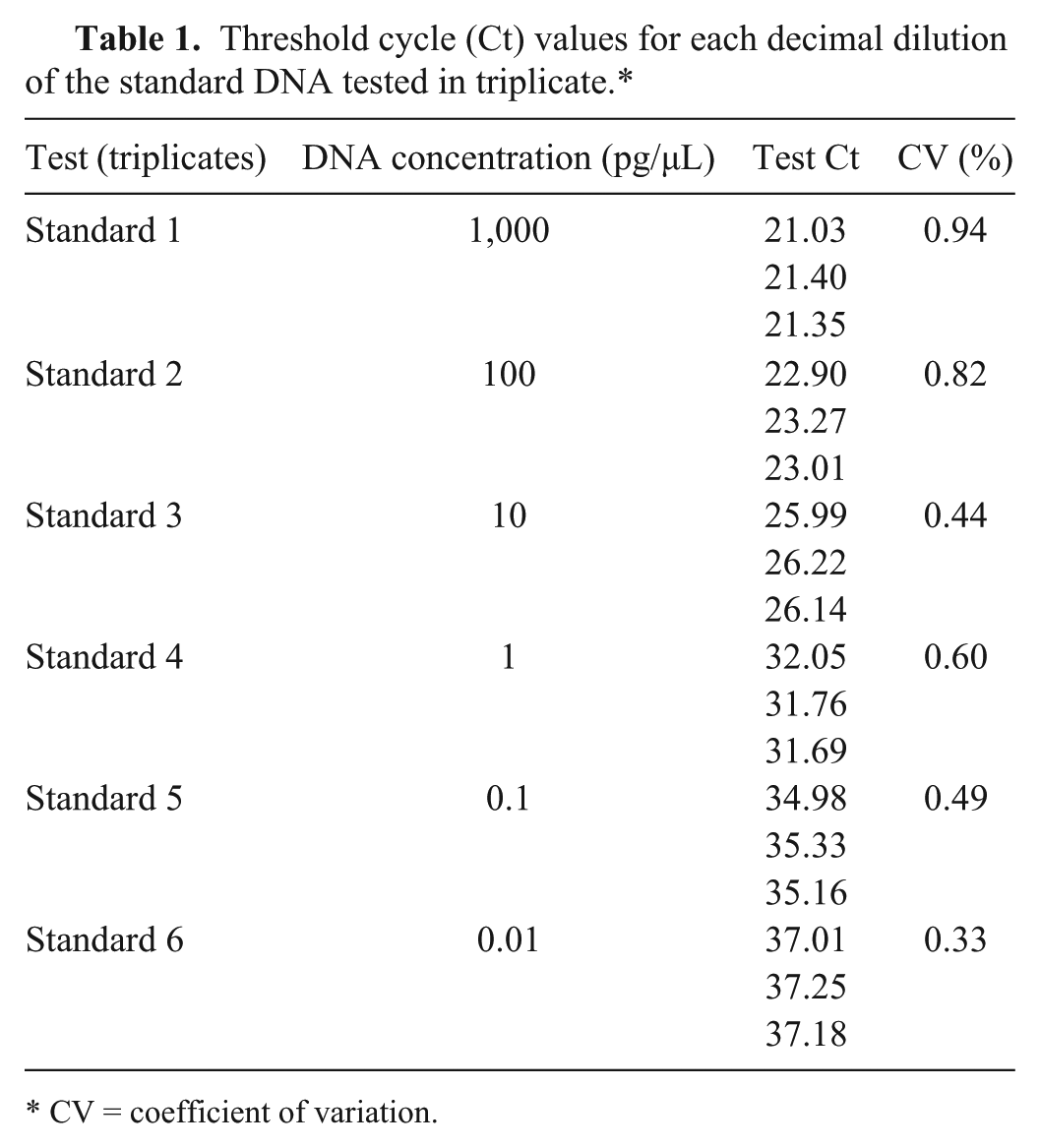
CV = coefficient of variation.
Repeatability was expressed by the correlation between values obtained from independent tests. Intra-assay repeatability was evaluated on 5 replicates of 7 positive and 7 negative DNA samples analyzed in a single assay run. Additionally, intra-assay repeatability was evaluated on 5 replicates of the 6 serial DNA dilutions used for LOD determination. Interassay repeatability was evaluated on the same DNA panel by 2 different operators in the same laboratory, in a short period of time (10 d), using the same materials and equipment over 20 runs. The AF-targeted real-time PCR interassay reproducibility was evaluated by running the test in 2 different laboratories (Ce.Tox and Molecular Biology Department) by analysis of the same panel of DNA samples in 20 runs.
Robustness was determined by applying small but deliberate variations to the protocol factors (Table 2) according to the fractional factorial design approach. 14 Genomic extracts from 8 positive samples were assayed in triplicate applying the controlled variations.
List of factors, with their suboptimal alternative values, included for robustness assessment.*
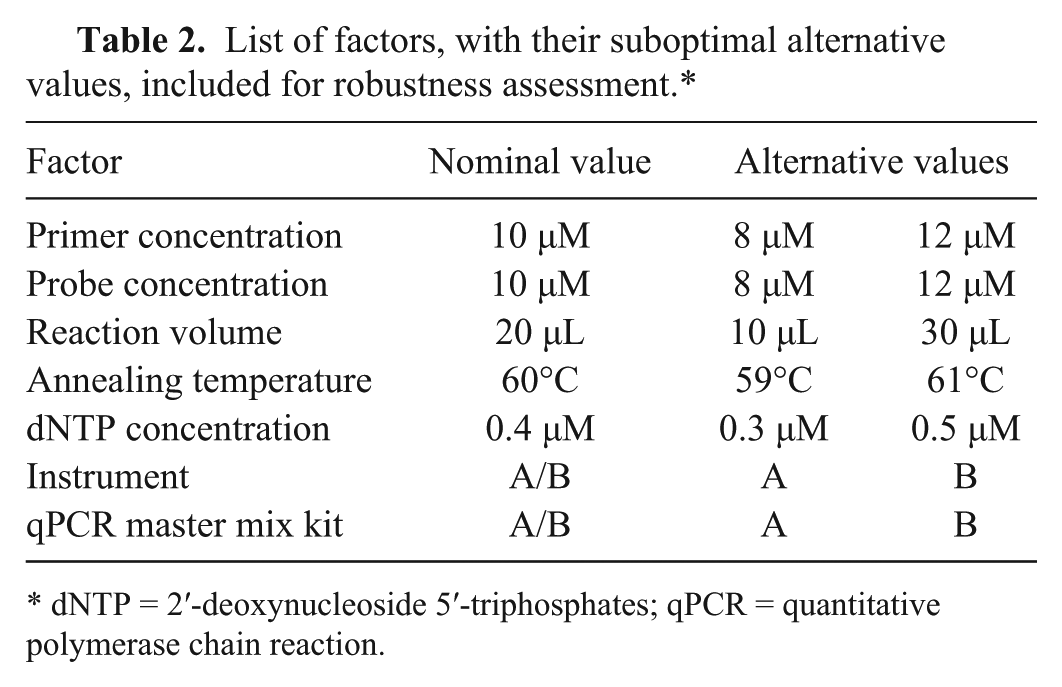
dNTP = 2′-deoxynucleoside 5′-triphosphates; qPCR = quantitative polymerase chain reaction.
A total of 120 positive and 120 negative results (30 positives and 30 negatives for each of 4 matrices, respectively) were confirmed by employing the real-time PCR. A good separation between the results of all negative and positive samples was recorded. Validation analyses resulted in a DSe and DSp of 100% indicating that all of the true-positive and true-negative samples were always correctly positive and negative according to the AF-targeted real-time PCR test. The LOD threshold was as low as 0.01 pg/μL. PCR amplification resulted in 92–109% efficiency, with a mean slope value of −3.3 ± 0.2. Results also demonstrated a high degree of precision, with a mean CV of 0.60% (0.33–0.94%) and R2 of 0.988 (Fig. 3).
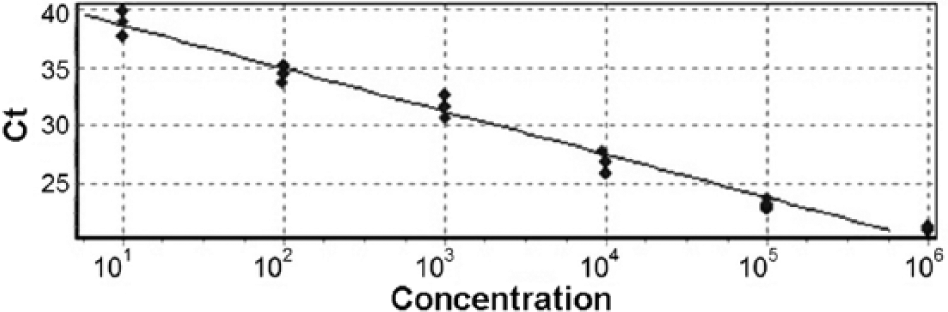
Standard curve generated by the analysis of triplicate 6-fold serial dilutions of Toxoplasma gondii DNA standard. Ct = threshold cycle.
The intra- and interassay repeatability was 100%. Reproducibility, evaluated through running the assay in the 2 laboratories, was 100% indicating that true-positive and true-negative samples were confirmed in different experimental conditions. The real-time PCR revealed a high level of robustness given that changing the assay factors had no influence on the overall results. The advantage of the AF real-time PCR is the rapidity and reliability of a semiautomated tool that limits the risk of cross-contamination as compared to the B1 nested PCR OIE protocol. LOD determination carried out with known amounts of target DNA demonstrated that quantities as low as 0.01 pg could be detected. This suggests that the TaqMan assay could also be used for quantitative analyses in epidemiologic studies.
Although our real-time PCR method does not provide evidence of cyst viability, it allows rapid detection of T. gondii in different biological matrices and it is sensitive and inclusive enough not to miss positive samples. This type of test could guide the enactment of public health measures and could be used to analyze raw meat and unprocessed meat products including those that are potential sources of Toxoplasma infection.
Footnotes
Authors’ contributions
AMF Marino contributed to conception and design of the study and critically revised the manuscript. RP Giunta and A Salvaggio contributed to acquisition, analysis, and interpretation of data, and critically revised the manuscript. M Percipalle contributed to design of the study and drafted the manuscript. G Caracappa and T Alfonzetti contributed to analysis of data and critically revised the manuscript. A Aparo contributed to analysis of data. S Reale contributed to conception and design of the study and drafted the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
ATCC 50174D, LGC Standards, Milan, Italy.
b.
Sigma-Aldrich, St. Louis, MO.
c.
Stomacher 80 biomaster, Sewar, Worthing, United Kingdom.
d.
Eppendorf UK, Stevenage, United Kingdom.
e.
Taq Gold 10X buffer, Taq Man genotyping master mix, Thermo Fisher Scientific, Waltham, MA.
f.
One-for-All extraction kit, Qiagen, Valencia, CA.
g.
BMR Genomics, Padua, Italy.
h.
Light Cycler TaqMan Master apparatus, Roche Diagnostics, Mannheim, Germany.
i.
Celbio Thermal Cycler One Advanced, Euroclone, Milan, Italy.
j.
Bio-Rad Laboratories, Hercules, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Grant funding for this study was awarded by the Italian Ministry of Health (Ricerca Corrente IZS SI 17/12/RC).