
Abstract
We compared the nested internal transcribed spacer (ITS) PCR and the 18S PCR-RFLP (restriction-fragment length polymorphism) pan-trypanosome assays in a cross-sectional survey of bovine trypanosomiasis in 358 cattle in Kwale County, Kenya. The prevalence of trypanosomiasis as determined by the nested ITS PCR was 19.6% (70/358) and by 18S PCR-RFLP was 16.8% (60/358). Of the pathogenic trypanosomes detected, the prevalence of Trypanosoma congolense and Trypanosoma vivax was greater than that of Trypanosoma simiae. The nested ITS PCR detected 83 parasite events, whereas the 18S PCR-RFLP detected 64; however, overall frequencies of infections and the parasite events detected did not differ between the assays (χ2 = 0.8, df = 1, p > 0.05 and χ2 = 2.5, df = 1, p > 0.05, respectively). The kappa statistic (0.8) showed good agreement between the tests. The nested ITS PCR and the 18S PCR-RFLP had comparable sensitivity, although the nested ITS PCR was better at detecting mixed infections (χ2 = 5.4, df = 1, p < 0.05).
Trypanosomiasis is a severe chronic wasting disease of domestic animals. Wildlife are also susceptible to infection with trypanosomes, 1 but they are less likely to succumb to the disease 17 and instead act as reservoirs. 14 The important animal trypanosomes include Trypanosoma congolense, Trypanosoma brucei brucei, Trypanosoma vivax, Trypanosoma evansi, Trypanosoma equiperdum, and Trypanosoma simiae. The geographic distribution of these trypanosomes is influenced by their modes of transmission (Table 1). Tsetse fly (Glossina spp.) transmission of trypanosomes is restricted to sub-Saharan Africa; the mechanically transmitted species occur beyond the tsetse belt. Mechanical transmission of T. congolense is also possible 5 ; however, this has not been confirmed in nature. With the varied modes of transmission and no vaccine to date, the control of animal trypanosomiasis remains a major challenge.
The distribution of pathogenic animal trypanosomes, affected animal hosts, and mode of transmission.

Animal trypanosomiasis causes low milk output, poor carcass quality, inability of animals to provide draught power, and high mortality rates in some cases. 10 Affected countries, in collaboration with international development agencies, have intervened by introducing various control measures, such as vector control, chemoprophylaxis and chemotherapy, and community education. The impact of these interventions requires periodic assessment through surveillance with a robust detection test. Documentation of the performance of detection tests in different geographic locations where animal trypanosomiasis is endemic should be encouraged in order to understand their feasibility as tools for surveillance of the disease.
Detection tests for animal trypanosomiasis include parasitologic, immunologic, and molecular methods, 7 but, for economic reasons, parasitologic techniques are still favored in sub-Saharan Africa despite their lack of sensitivity and labor intensiveness. Undoubtedly, PCR is the most appropriate tool for sensitive screening of animal trypanosomiasis and retrospective analysis. Species-specific PCRs require multiple reactions per sample to detect the different species of trypanosomes, 11 which is overwhelming during large-scale operation. Thus, pan-trypanosome PCRs are the preferred assays for epidemiologic survey targeting multiple species of trypanosomes.3,6,8,15
The ribosomal (r)DNA-based pan-trypanosome PCRs (Table 2) are commonly used for detection of animal trypanosomes; however, only a few of these assays have been comparatively evaluated on field samples.15,18 The nested internal transcribed spacer (ITS) PCR and the 18S PCR-RFLP (restriction-fragment length polymorphism) pair lacks such comparative data. Thus, we compared the 2 assays on field samples obtained from Kwale County, Kenya, where trypanosomiasis is endemic. Kwale County is covered with dense vegetation and all-season streams, consequently providing the required humidity for proliferation of the vectors. The occurrence of the various species of animal trypanosomes in the region has been reported,12,16 thus making the region attractive for evaluation of these tests. Additionally, it was important to understand the current burden of bovine trypanosomiasis in the region as a follow-up to ongoing control efforts.
Different ribosomal (r)DNA-based pan-trypanosome PCR tests for African trypanosomiasis.

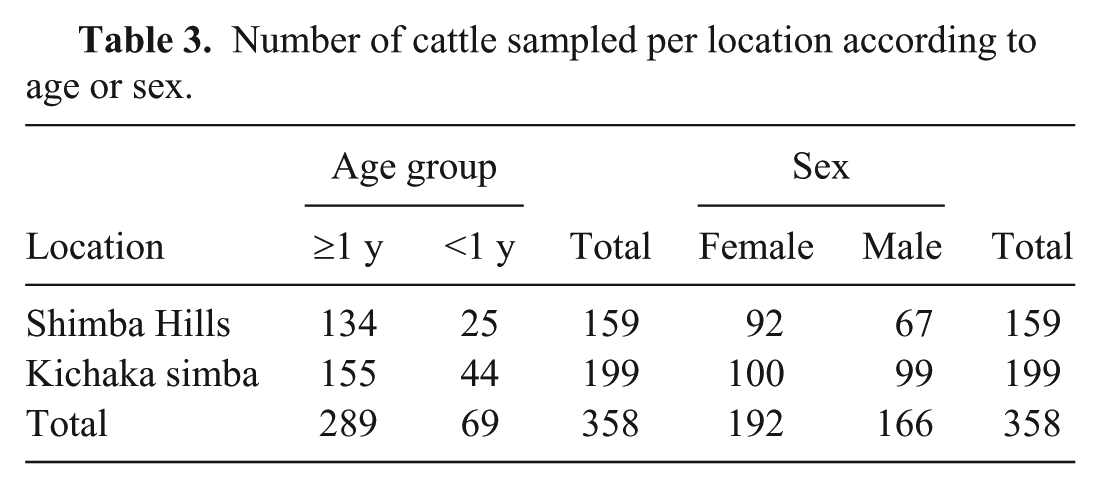
Reference DNA samples were prepared from T. congolense savannah (TC13; courtesy of Dr. Henry Tabel, Canada), T. b. brucei (AnTat1.1E), T. vivax (ILRAD 700), and T. evansi (STIB816, obtained from the Institute of Tropical Medicine, Antwerp). The trypanosome strains were propagated in mice (C57BL/6) and harvested when parasitemia was 1 × 107/mL. Sampling was conducted in 2 villages, namely Shimba Hills (4°21′16.8″S and 39°24′54.2″E) and Kichaka simba (4°20′59.35″S and 39°21′33.61″E) in May 2013 (Table 3). During sampling, cattle were restrained in a crush pen, and whole blood was collected by the jugular route into EDTA-coated tubes. a The animal experiments were approved by the Ethical Committee for Animal Experiments of the Vrije Universiteit Brussel (clearance number 11-220-6).
Number of cattle sampled per location according to age or sex.
DNA was extracted from whole blood (10 µL) using a commercial kit b ; the concentration was determined using a spectrophotometer, c and samples were stored at −20°C until analyzed. The samples were examined for trypanosome DNA by the nested ITS PCR as described previously. 3 Briefly, the first run consisted of 5 µL of the template, 1× Taq DNA polymerase buffer, d 0.2 mM of each dNTP, 0.4 µM of each primer (ITS1 and ITS2), 1.25 U Taq DNA polymerase, e and adjusted with distilled water to 25 µL/reaction. For the second run, the product of the first run (1 µL) was seeded in 24 µL of a fresh master mix prepared as above except the ITS3 and ITS4 primers were used in the place of ITS1 and ITS2. The cycle conditions were the following: lid temperature 105°C; initial denaturation 95°C for 5 min; 94°C for 30 s, 57°C for 30 s, 72°C for 60 s for 35 cycles; postcycle 72°C for 10 min; and paused at 12°C. The amplified products of the second run were separated by electrophoresis on 1.5% (w/v) agarose f at 100 V for 40 min. The profiles on gels were scored according to the standard profiles obtained from reference trypanosomes and a previous study. 3
The detection of trypanosome DNA using the 18S PCR-RFLP was performed as described. 8 Briefly, the master mix was prepared from 1× Taq DNA polymerase buffer, 0.2 mM of each dNTP, 0.5 µM forward primer 18ST nF2 and 0.5 µM reverse primer 18ST nR3 for the first PCR, or 0.5 µM reverse primer 18ST nR2 for the semi-nested PCR and 1.25 U Taq DNA polymerase. The master mix was adjusted with distilled water to the required volume. Whereas the first PCR master mix (20 µL) was seeded with an extracted DNA sample (5 µL), the second PCR master mix (24 µL) was seeded with the first PCR product (1 µL). The cycle conditions were the following: lid temperature 105°C; precycle 95°C for 4 min; 92°C for 30 s, 58°C for 45 s, 72°C for 60 s for 40 cycles; postcycle 72°C for 10 min; and paused at 4°C. Amplified products of the second run were separated by electrophoresis on 2% (w/v) agarose. PCR amplicons of ±600 bp were double digested with the restriction enzymes MspI g and Eco57I. h For the digestion, 4 µL of the PCR product were added to 11 µL of premix containing 2.4 U MspI, 2.4 U Eco57I, 1× Cut smart buffer, i 1× SAM buffer, j and 8.6 µL of distilled water. The mixture was incubated overnight at 37°C, and the digested products were separated by electrophoresis on 2% (w/v) agarose gel at 100 V for 40 min. The profiles were interpreted according to the RFLP profiles obtained from the reference strains and as reported in previous studies.4,8
Conflicting profiles were cross-validated through sequencing of the PCR products. For this, 10 µL of the positive PCR products were processed by incubating with 2 U alkaline phosphatase k and 20 U exonuclease I l on a thermal cycler m programmed at 37°C for 15 min and then at 80°C for 15 min. The sequencing of the processed sample was outsourced to a VIB genetic service facility (University of Antwerp). Homology search in the databank was performed using a bioinformatic software. n
Statistical analysis was performed, o and frequencies were compared using the chi-square (χ2) test. Chosen confidence level (α) was 0.05, and p values <0.05 were considered significant. The level of agreement between the 2 tests was estimated using kappa statistics. 13
The prevalence of animal trypanosomiasis by the nested ITS PCR was 19.6% (70/358). The nested ITS PCR detected T. congolense Savannah 2% (7/358), T. congolense Kilifi 3.4% (12/358), T. vivax 4.7% (17/358), T. simiae 1.4% (5/358), T. theileri 4.7 % (17/358), T. congolense Savannah/T. vivax mixed 0.3% (1/358), T. congolense Kilifi/T. vivax mixed 0.6% (2/358), T. congolense Kilifi/T. theileri mixed 0.3% (1/358), T. vivax/T. simiae mixed 2% (7/358), and T. congolense Kilifi/T. vivax/T. simiae mixed 0.3 % (1/358; Table 4).The 18S PCR-RFLP was positive in 16.8% (60/358) of the samples, and detected T. congolense Savannah 2.2% (8/358), T. congolense Kilifi 3.9% (14/358), T. vivax 4.5% (16/358), T. simiae 0.6% (2/358), T. theileri 4.7 % (17/358), T. vivax/T. simiae mixed 0.6% (2/358), and T. congolense Kilifi/T. vivax/T. simiae mixed 0.3 % (1/358; Table 5). There was no significant difference in rates of positive samples detected between the 2 tests (χ2 = 0.8, df = 1, p > 0.05), and kappa statistics showed a substantial agreement (Table 6; kappa = 0.8). However, the nested ITS PCR detected more mixed infections than the 18S PCR-RFLP (χ2 = 5.4, df = 1, p < 0.05).
Single and mixed trypanosome infections detected with the nested internal transcribed spacer (ITS) PCR according to location, animal sex, or age group.*
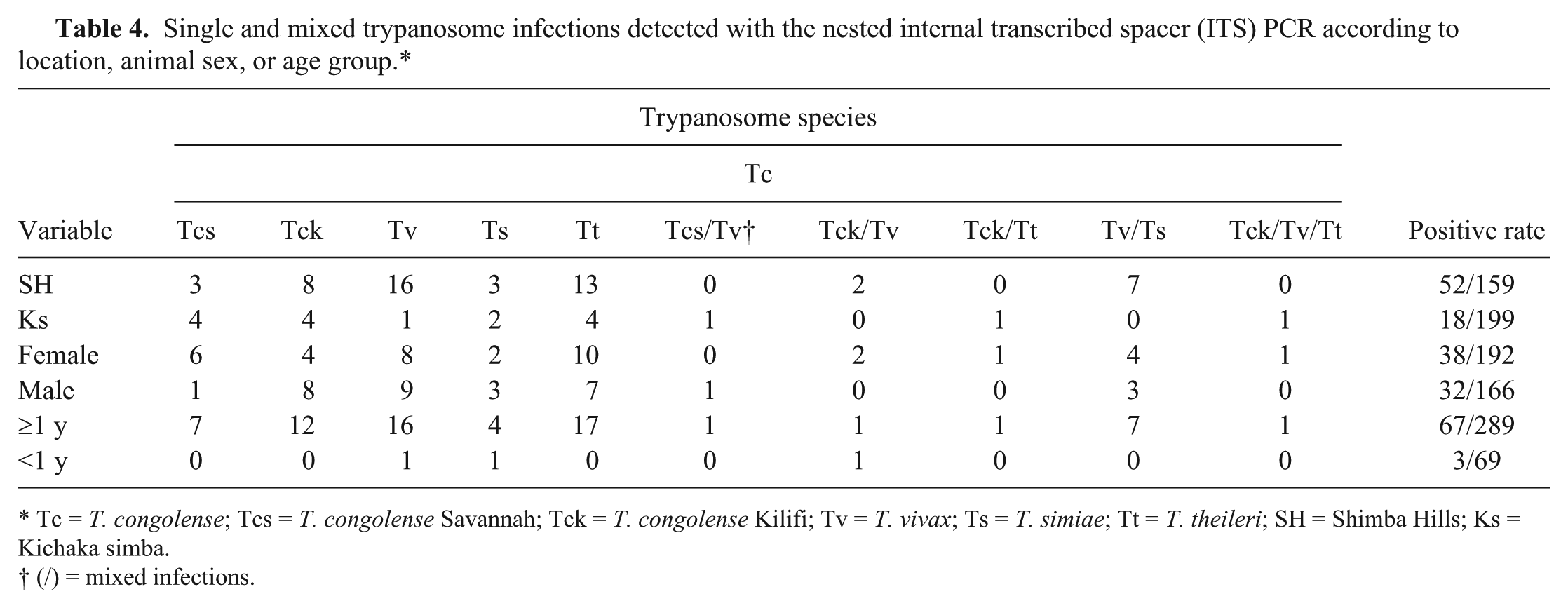
Tc = T. congolense; Tcs = T. congolense Savannah; Tck = T. congolense Kilifi; Tv = T. vivax; Ts = T. simiae; Tt = T. theileri; SH = Shimba Hills; Ks = Kichaka simba.
(/) = mixed infections.
Single and mixed trypanosome infections detected with the 18S PCR-RFLP (restriction-fragment length polymorphism) according to location, animal sex, or age group.*
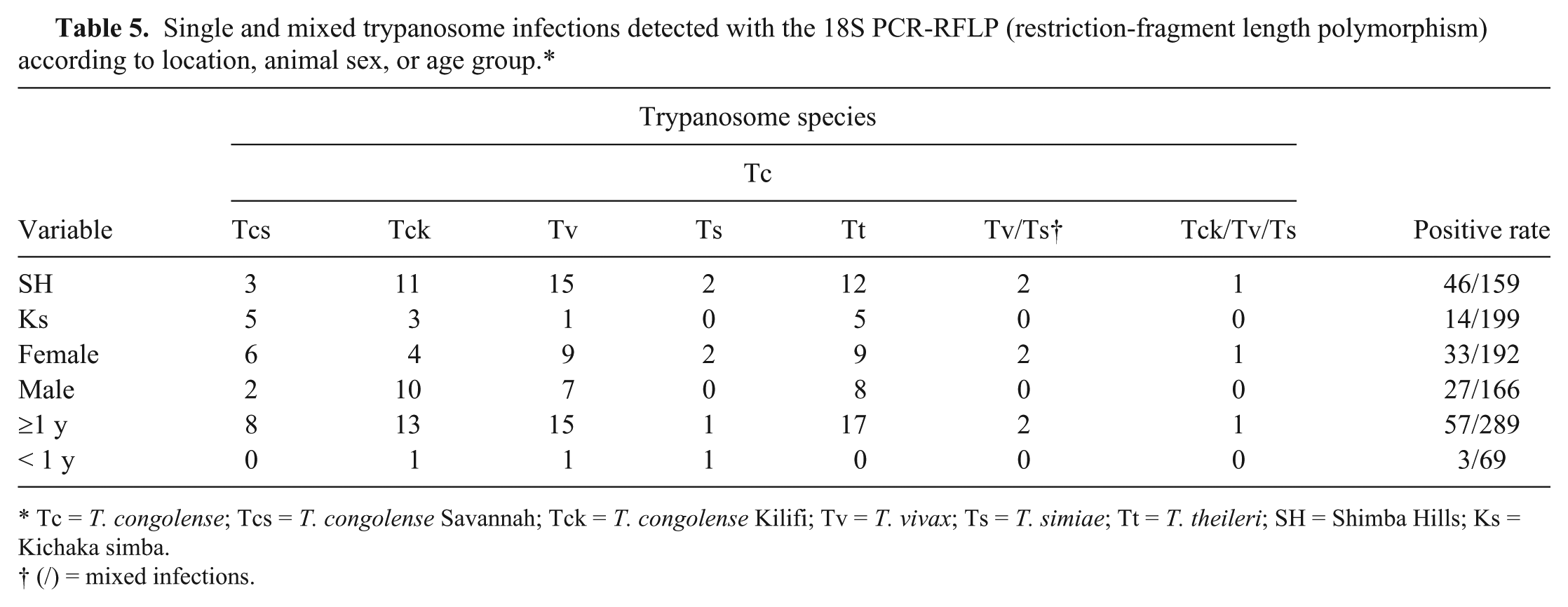
Tc = T. congolense; Tcs = T. congolense Savannah; Tck = T. congolense Kilifi; Tv = T. vivax; Ts = T. simiae; Tt = T. theileri; SH = Shimba Hills; Ks = Kichaka simba.
(/) = mixed infections.
The agreement between the nested internal transcribed spacer (ITS) PCR and the 18S PCR-RFLP (restriction-fragment length polymorphism).*

Kappa = 0.8; p < 0.05.
The nested ITS PCR detected 83 parasite events, whereas the 18S PCR-RFLP detected 64, but the difference was not significant (Table 7; χ2 = 2.5, df = 1, p > 0.05). We compared the frequency with which each of the assays detected the important species of trypanosomes (T. congolense, T. vivax, and T. simiae) in order to assess if either test was better at detecting a particular species. The nested ITS PCR and the 18S PCR-RFLP detected T. congolense (Savannah and Kilifi), T. vivax, and T. simiae at similar levels (χ2 = 0.02, df = 1, p > 0.05; χ2 = 1.72, df = 1, p > 0.05; and χ2 = 2.9, df = 1, p > 0.05, respectively). We assessed the predominant species responsible for bovine trypanosomiasis in the region and found that their frequencies varied significantly (χ2 = 14.8, df = 4, p < 0.05). T. congolense and T. vivax occurred at the same frequency (χ2 = 0.07, df = 1, p > 0.05). When the frequencies of T. congolense (Savannah and Kilifi) and T. vivax were compared to T. simiae, there were significant differences (χ2 = 5.8, df = 1, p < 0.05 and χ2 = 7.05, df = 1, p < 0.05, respectively). Also T. congolense Kilifi was more prevalent than T. congolense Savannah (χ2 = 4.5, df = 1, p < 0.05).
Events of the different species (subspecies) of trypanosomes detected by the nested internal transcribed spacer (ITS) PCR and the 18S PCR-RFLP (restriction-fragment length polymorphism).*

In both assays, infections encountered were caused by T. congolense (Savannah and Kilifi), T. vivax, T. simiae, and T. theileri, which occurred as single and mixed infections. The nested ITS PCR would detect mixed infections and the 18S PCR-RFLP would score positive on the same sample but only revealed single parasite infections. Failure of the 18S PCR-RFLP to detect a coinfecting species in mixed infections might be attributed to low amounts of DNA or obstruction of the profile by a coinfecting species. 4 From this observation, mixed infections are more likely to be missed with the 18S PCR-RFLP than with the nested ITS PCR. The nested ITS PCR could clearly differentiate all species; however, the profiles of the 2 T. congolense Savannah and Kilifi subspecies were conflicting. To clearly distinguish the nested ITS PCR profiles of T. congolense Savannah from Kilifi, the PCR products were separated for 50 min at 100 V and in parallel with the reference strain samples. In addition, sequencing of the PCR product and cross-referencing with the 18S PCR-RFLP results resolved the conflicts between the 2 T. congolense subspecies. We also observed that the 18S PCR-RFLP could not readily differentiate T. theileri from T. simiae as was reported previously. 8 The 2 species were differentiated by sequencing the PCR products and cross-referencing with the nested ITS PCR results. Alternatively, T. theileri and T. simiae amplified by the 18S PCR-RFLP can be differentiated by digesting the PCR product using the MobII restriction enzyme. 8
Of these 3 pathogenic trypanosomes (T. congolense, T. vivax, and T. simiae), analysis of frequencies showed no difference between T. vivax and T. congolense; the frequency of T. simiae was lower than T. congolense and T. vivax. The detection of fewer T. simiae compared to T. congolense or T. vivax was not surprising as both species are principal pathogens of cattle unlike T. simiae, which is more commonly encountered in pigs. 9 The number of cattle infected with T. simiae was considerable, yet recent studies12,16 did not report the presence of this trypanosome in the region where we conducted the study. The occurrence of T. simiae in cattle could be attributed to transmission of infections from infected wild pigs to cattle considering the location of the study sites, which are in the vicinity of Shimba Hills National Reserve. Previous studies employed parasitologic techniques; therefore, failure to identify T. simiae could possibly be the result of parasite loads below the detection limit of the technique or misclassification of the parasite. Analysis of T. congolense at the subspecies level revealed a higher frequency of T. congolense Kilifi than Savannah. This could be attributed to the fact that cattle might tolerate higher levels of the less pathogenic trypanosomes, 2 and this might provide the same explanation for the high prevalence of T. vivax. Where the circulating strain of trypanosomes is less virulent, infected animals will not readily succumb or manifest signs that would alert their owners. This promotes accumulation of less virulent species to a higher frequency compared to the more virulent species that are highly likely to be detected and treated.
We recommend the use of the nested ITS PCR for detection of trypanosomes as it proved to be a sensitive technique for detection of trypanosomes occurring in both single and mixed infections. Practically, the inability of the assay to classify certain species (subspecies) is of less importance if the aim of detection is therapeutic intervention because these trypanosomes are treated with the same drugs. The methodological disadvantage of the 18S PCR-RFLP is the additional step that involves digestion of amplified product, which makes the assay time consuming and costly, and affects its sensitivity in cases where the concentration of amplified product is low. Nonetheless, unique restriction patterns provided by the 18S PCR-RFLP allow easy characterization of trypanosomes because only a few species have conflicting profiles by this assay.
Footnotes
Acknowledgements
We appreciate the valuable contributions from Dr. Hamisi Mwalonya, Kwale County Veterinary Officer; Dr. George Adinoh Omondi, Dawn Maranga, and Thomas Adinoh who are all research assistants at the Institute of Primate Research (Nairobi); and Nancy Nyambura Ndumia who was an intern in the laboratory of Prof. Dr. ir. Stefan Magez. Finally, we acknowledge Dr. ir. Stijlemans Benoît for critically reviewing the entire manuscript.
Authors’ note
S Odongo is a research fellow at Vrije Universiteit Brussel.
Authors’ contributions
S Odongo contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; and drafted the manuscript. V Delespaux contributed to acquisition, analysis, and interpretation of data. M Ngotho and S Magez contributed to conception and design of the study, and contributed to acquisition, analysis, and interpretation of data. SM Bekkele contributed to acquisition, analysis, and interpretation of data, and drafted the manuscript. All authors critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Vacutainer tube EDTA (K2) 6 mL, BD, Franklin Lakes, NJ.
b.
DNeasy blood and tissue (50), Qiagen GmbH, Hilden, Germany.
c.
NanoDrop, Thermo Fisher Scientific, Wilmington, DE.
d.
GoTaq buffer, Promega Corp., Madison, WI.
e.
GoTaq DNA polymerase, Promega Corp., Madison, WI.
f.
SeaKem LE agarose, Lonza Inc., Rockland, ME.
g.
MspI, New England Biolabs Inc., Leiden, The Netherlands.
h.
Eco57I, Thermo Fisher Scientific Inc., Erembodegem–Aalst, Belgium.
i.
CutSmart buffer, New England Biolabs Inc., Leiden, The Netherlands.
j.
SAM buffer, Thermo Fisher Scientific Inc., Erembodegem–Aalst, Belgium.
k.
FastAP, Thermo Fisher Scientific Inc., Erembodegem–Aalst, Belgium.
l.
Exonuclease I, Thermo Fisher Scientific, Erembodegem–Aalst, Belgium.
m.
MWG BIOTECH Primus 96plus 220 VAC, Cole-Parmer, Belgium.
n.
Clone Manager Professional 9, Sci-Ed Software, Morrisville, NC.
o.
SPSS16 statistical program, Polar Engineering and Consulting, Nikiski, AK.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the EU/FP7 HEALTH 2007-2.3.4-1 NANOTRYP project.