
Abstract
African swine fever (ASF), classical swine fever (CSF), and foot-and-mouth disease (FMD) are highly contagious animal diseases of significant economic importance. Pigs infected with ASF and CSF viruses (ASFV and CSFV) develop clinical signs that may be indistinguishable from other diseases. Likewise, various causes of vesicular disease can mimic clinical signs caused by the FMD virus (FMDV). Early detection is critical to limiting the impact and spread of these disease outbreaks, and the ability to perform herd-level surveillance for all 3 diseases rapidly and cost effectively using a single diagnostic sample and test is highly desirable. This study assessed the feasibility of simultaneous ASFV, CSFV, and FMDV detection by multiplex reverse transcription real-time polymerase chain reaction (mRT-qPCR) in swine oral fluids collected through the use of chewing ropes. Animal groups were experimentally infected independently with each virus, observed for clinical signs, and oral fluids collected and tested throughout the course of infection. All animal groups chewed on the ropes readily before and after onset of clinical signs and before onset of lameness or serious clinical signs. ASFV was detected as early as 3 days postinoculation (dpi), 2–3 days before onset of clinical disease; CSFV was detected at 5 dpi, coincident with onset of clinical disease; and FMDV was detected as early as 1 dpi, 1 day before the onset of clinical disease. Equivalent results were observed in 4 independent studies and demonstrate the feasibility of oral fluids and mRT-qPCR for surveillance of ASF, CSF, and FMD in swine populations.
Introduction
African swine fever (ASF), classical swine fever (CSF), and foot-and-mouth disease (FMD) are highly contagious transboundary animal diseases that pose grave and catastrophic economic consequences to agriculture. Because of their potential for rapid spread, these diseases pose risks to trading partners, further impacting international trade and making them reportable to the World Organization for Animal Health (OIE). ASF-, CSF-, and/or FMD-free countries maintain vigilance through passive and/or active disease surveillance to ensure early detection to mitigate the spread and impact of potential outbreaks of disease and to maintain trading confidence with international partners.
Although ASF and CSF are caused by unrelated viruses belonging to different DNA and RNA virus families, Asfarviridae and Flaviviridae, respectively, both have similar systemic clinical signs, such as fever, ataxia, and severe depression. In this regard, the clinical signs of ASF and CSF can be similar to domestic swine diseases including porcine circovirus-associated diseases, erysipelas, pasteurellosis, streptococcosis, and salmonellosis, thus possibly delaying detection.27,28 Likewise, detection of the FMD virus (FMDV), a small positive-sense RNA virus belonging to family Picornaviridae, may be delayed because it causes vesicular disease that is visibly indistinguishable from other less contagious vesicular diseases in pigs and other cloven-hoofed animals. 29 Early detection is critical to limiting the impact and spread of these diseases, and the ability to perform surveillance for all 3 diseases rapidly and cost effectively using a single diagnostic sample and test is highly desirable.
Swine oral fluid (OF) has emerged as a highly effective and convenient herd-level sample for disease surveillance. Detection of nucleic acid and antibodies for Porcine reproductive and respiratory syndrome virus (PRRSV), Porcine circovirus-2 (PCV-2), Influenza A virus, Mycoplasma hyopneumoniae, and torque teno virus in OF samples have been reported (Vansickle J. Oral fluid collection enhances testing. National Hog Farmer 2010 September 22. Available from: http://nationalhogfarmer.com/health-diseases/disease-prevention/oral-fluid-collection-enhances-testing-0915).2,9,14–17,19 In 2013, studies reported the experimental detection of FMDV genome or ASF virus (ASFV) antibody in swine OF samples.13,24
Specifically, OF is defined as the fluid in the oral cavity collected by use of an absorptive device. 18 OF samples are collected by suspending a cotton rope in a pig pen and allowing the pigs to chew on the rope for 30 min. OF is easily collected by squeezing the rope. As naturally curious and social creatures, pigs will, within 10–20 min, investigate and saturate ropes with OF, significantly relieving the burden of individual animal sampling. 11 Because pigs continually orally investigate their environment, this approach provides a combined sample of both individual animals and their environment.
Oral fluid samples include secretions from salivary glands, upper gastrointestinal and respiratory tracts, and the gingival sulcus. Serum components, including antibodies, hormones, bacteria, and viruses, are present in OF by their transfer through capillary walls in salivary glands and transport via gingival fluids and mucosal cells. 30 The oropharynx is an important site for FMDV replication. 22 In addition, ASFV and CSFV replication occur in the tonsil and pharyngeal region resulting in virus secretions from the nasal pharyngeal5,6,8,31 and oropharyngeal compartments. 26 All 3 disease agents are highly infectious and may be spread by direct or indirect contact via oronasal and oropharyngeal routes.
To investigate the potential use of swine OF as a herd sample for combined and streamlined detection of ASFV, CSFV, and FMDV, the present study focused on development of an optimized RNA and DNA detection workflow based on multiplex reverse transcription real-time polymerase chain reaction (mRT-qPCR). The feasibility of OF sampling for ASF, CSF, and FMD was assessed individually in experimentally infected animals, and performance of the nucleic acid purification and mRT-qPCR workflow was assessed using rope-collected OF samples.
Materials and methods
Reference strains
Viruses used for the analytical sensitivity analysis were as follows: ASFV Kimakia-64 (3.3 × 107 TCID50/mL), CSFV San Cristobal A tons (5.2 × 108 TCID50/mL), and FMDV A Iran 1/98 (6.6 × 105 TCID50/mL); 50 µL of each virus solution was used for nucleic acid purification. The ASFV Georgia 2007/1 strain was provided by the Department of Homeland Security Science and Technology Directorate, Plum Island Animal Disease Center (PIADC; Plum Island, New York) through a kind contribution from Drs. Vepkhvadze and Donduashvili at the Laboratory of the Ministry of Agriculture, Tbilisi, Georgia. ASFV (Kimakia-64 and Lisbon 60 strains), CSFV (San Cristobal A tons, Haiti-96, and Brescia strains), and FMDV (A Iran 1/98 and O1 Brugge iso79 serotypes) were obtained from the microbial repository at the U.S. Department of Agriculture, National Veterinary Services Laboratories’ Foreign Animal Disease Diagnostic Laboratory (USDA-NVSL-FADDL; Plum Island, New York).
Experimental infections and sample collection
All animal experiments involving ASFV, CSFV, or FMDV were carried out at the PIADC according to Institutional Animal Care and Use Committee protocols. Animal studies were performed for the primary purpose of demonstration of hallmark clinical disease for FMD, CSF, and ASF associated with the USDA Foreign Animal Disease Diagnostician School, and rope sampling for OF was incorporated into multiple independent animal inoculation studies to obtain samples that enabled leveraging of resources. Four independent studies were performed months apart (Table 1). The virus dose and routes of inoculation were specific to each virus study group and were replicated in the 4 independent studies for each respective virus. Yorkshire pigs (2–3 months old, weighing 23 kg) were housed in separate rooms by disease, and experimentally inoculated with ASFV, CSFV, or FMDV.
Experimental viral infections and study group design for oral fluid sampling.*
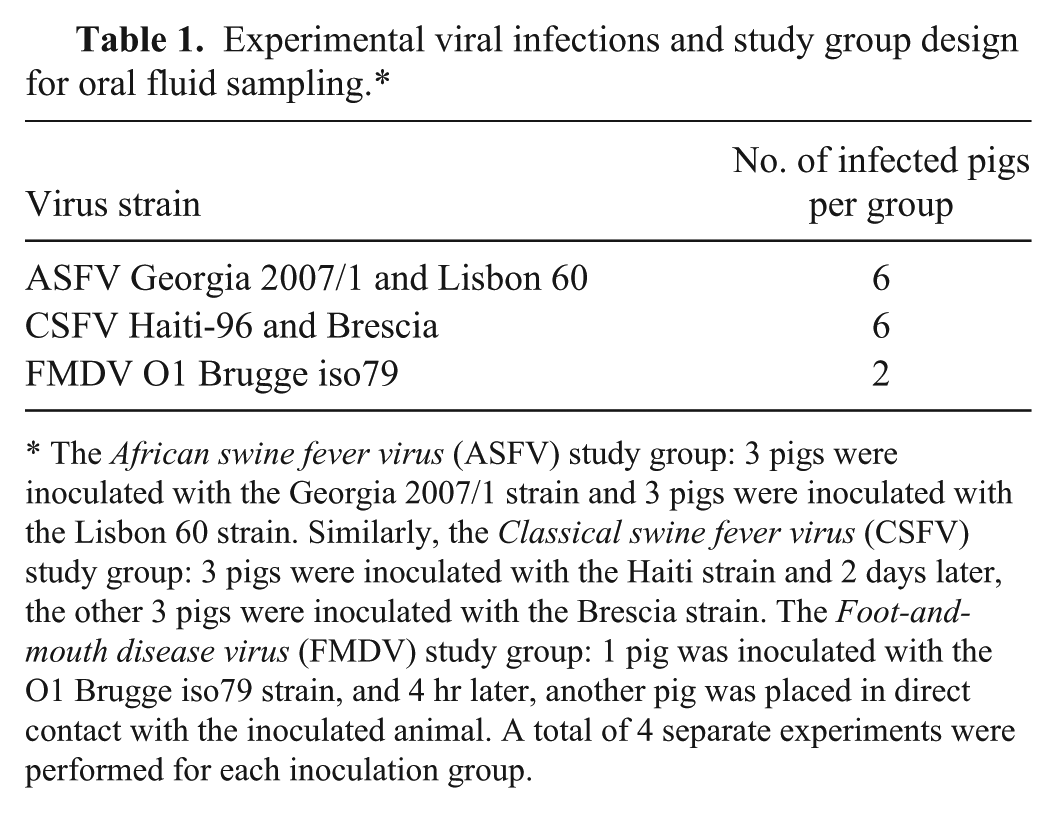
The African swine fever virus (ASFV) study group: 3 pigs were inoculated with the Georgia 2007/1 strain and 3 pigs were inoculated with the Lisbon 60 strain. Similarly, the Classical swine fever virus (CSFV) study group: 3 pigs were inoculated with the Haiti strain and 2 days later, the other 3 pigs were inoculated with the Brescia strain. The Foot-and-mouth disease virus (FMDV) study group: 1 pig was inoculated with the O1 Brugge iso79 strain, and 4 hr later, another pig was placed in direct contact with the inoculated animal. A total of 4 separate experiments were performed for each inoculation group.
For the ASFV-infected group, 6 pigs were housed per room; 3 pigs were inoculated with 1 mL of viral suspension by intramuscular injection (in the neck) with the Georgia 2007/1 strain (titer = 103 TCID50/mL), and 3 pigs were inoculated with the Lisbon 60 strain (titer = 103 TCID50/mL). For the CSFV-infected group, 6 pigs were housed per room; 3 pigs were inoculated with 1 mL of viral suspension via intramuscular injection (in the neck) containing the CSFV low virulence Haiti strain (titer = 103 TCID50/mL), and, 2 days later, the remaining 3 pigs were inoculated intranasally with 1 mL of viral suspension per nostril with the CSFV high-virulence Brescia strain (titer = 103 TCID50/mL). For the FMDV-infected group (n = 2), 1 pig was inoculated in the left rear heel bulb with 1.0 mL of FMDV O1 Brugge strain (titer = 106 TCID50/mL). A second pig was placed in the room, in direct contact with the inoculated animal, 4 hr after inoculation.
For the collection of OF, a single 3.81 cm diameter by 1.22 m length cotton rope with 4 interwoven strands was used per pen per room. The rope was hung on the wall, above the ground, at pigs’ shoulder height. Rope strands were unraveled to allow multiple pigs to chew on the rope simultaneously. A clean rope was hung for 20–30 min in the morning, prior to feeding, daily, for the duration of each animal study. Chewed ropes were manually wrung out into plastic bags, aliquoted into 1-mL fractions and stored at −70°C. Animals with severe clinical signs were euthanized as needed throughout the study. Oral swabs (4 per animal) were collected, pooled, and resuspended in 12 mL of Tris buffered tryptose broth (TBTB) medium. a
Four independent experimental infections with incorporated OF sampling were performed for ASFV, CSFV, and FMDV resulting in 4 study groups for each pathogen. However, because the OF sampling was a leveraged study, experimental design and animal access were limited, resulting in nonmatched days postinoculation (dpi) OF collection time points for the 4 studies. Furthermore, the standard sample(s) for detection of each pathogen (i.e., blood for ASFV; nasal swabs, tonsil scrapings, and blood samples for CSFV; and epithelium, esophageal–pharyngeal samples and serum for FMDV) were not collected given the limitations of animal use protocols. Additionally, the limit of detection (number of positive animals that can be detected among the total number of animals in the pen), an important performance parameter of oral fluids, was not possible to assess in this study.
Plasmids and control RNA
DNA sequences, specific to ASFV, CSFV, and FMDV assay target sequences, were synthesized and cloned into plasmid vectors by a commercial company. b In vitro transcription of a CSFV and FMDV fusion construct was performed using a commercial kit according to the manufacturer’s instructions. c In vitro transcripts and plasmids were quantified using a spectrophotometer, d and sizes were verified by electrophoresis. e An exogenous internal PCR control RNA (XIPC) was prepared as described previously. 21 The positive amplification control (PAC) was prepared by combining ASFV target containing plasmid DNA with in vitro transcribed CSFV, FMDV, and XIPC target RNA at 1,000 copies/µL each. PAC was used for each mRT-qPCR run to ensure functionality of the mRT-qPCR conditions. The XIPC was also spiked into the nucleic acid purification of test samples to monitor the efficacy of the nucleic acid purification reagents and methods.
Oligonucleotides
Sequence information and the final reaction concentrations for oligonucleotides employed in the mRT-qPCR assay are provided in Table 2. Primer and TaqMan probe sequences for detection of ASFV, CSFV, FMDV, and XIPC were taken from previous publications,1,4,20,21 and optimal oligonucleotide concentrations were determined through empirical testing. Fluorescence and/or quencher molecules for oligonucleotide probes for ASFV and FMDV were modified from their original USDA designs in order to make the mRT-qPCR compatible for simultaneous discrimination of 4 different targets. All primers and probes were purchased from a commercial source. f Oligonucleotide concentrations were verified using a spectrophotometer d according to instructions in the RNA-to-CT 1-step kit manual. c
African swine fever virus (ASFV), Classical swine fever virus (CSFV), Foot-and-mouth disease virus (FMDV), and exogenous internal positive control (XIPC) reverse transcription real-time polymerase chain reaction primers and probes sequences, amplicon sizes, and oligonucleotide concentrations.
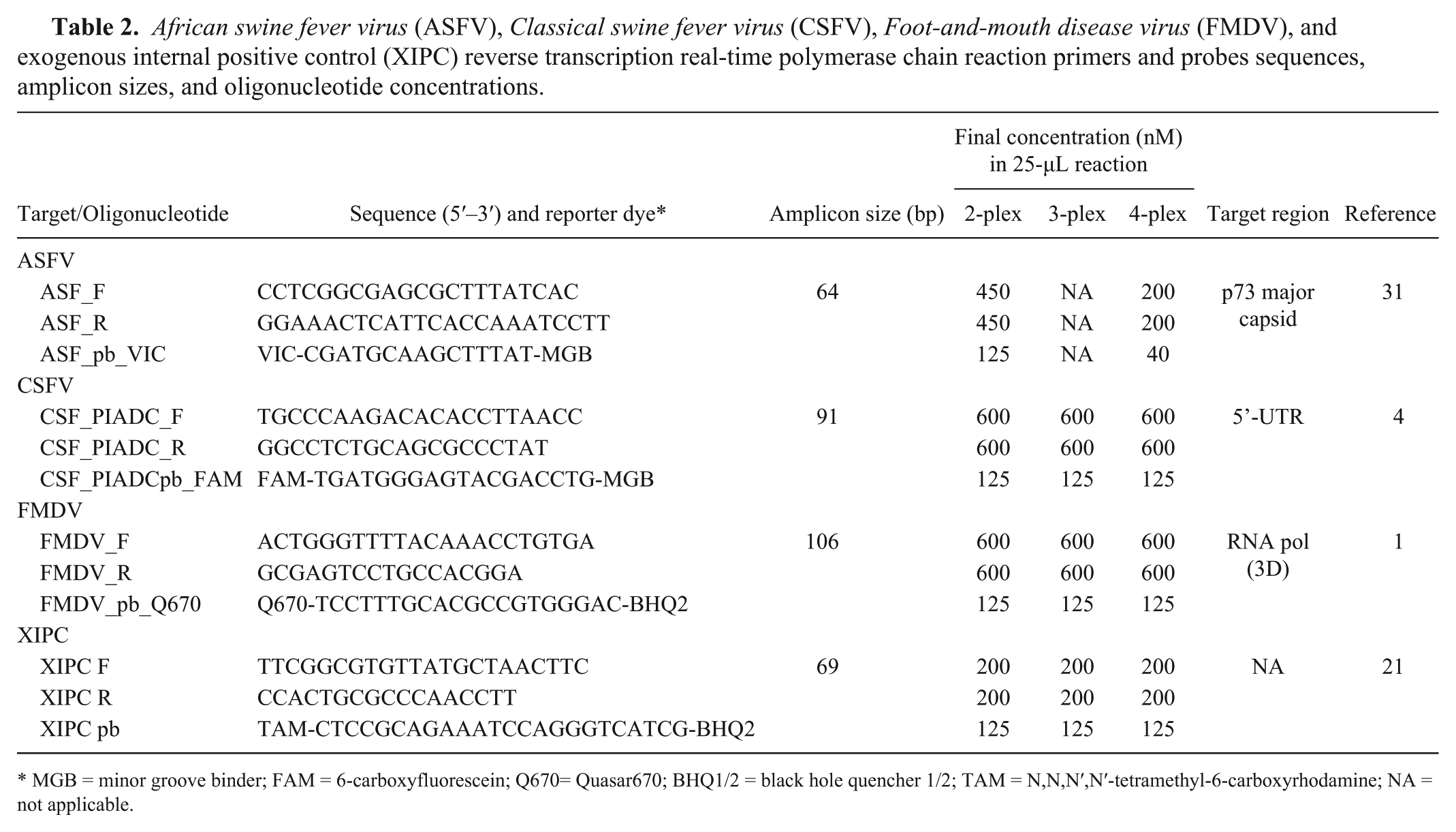
MGB = minor groove binder; FAM = 6-carboxyfluorescein; Q670= Quasar670; BHQ1/2 = black hole quencher 1/2; TAM = N,N,N′,N′-tetramethyl-6-carboxyrhodamine; NA = not applicable.
Nucleic acid purification
Oral fluid samples were initially centrifuged at 300 × g for 2 min to remove dirt and debris prior to nucleic acid extraction. After centrifugation, 250 µL of OF was added to 400 µL of lysis solution (containing 400 µL of lysis binding concentrate, c 1 µL of carrier RNA [1 µg/µL], and 1 µL of XIPC RNA [at 10,000 copies/µL]), mixed for 5 min at 15 Hz using a tissue lyser, e and centrifuged at 20,000 × g for 2 min to generate the clarified lysate. These sample clarification steps were performed using 1.5-mL microcentrifuge tubes, adjustable multichannel pipettes, and 8 × 12 array tube racks, which enabled easy, fast, and concurrent multiple sample processing. Nucleic acid was purified using a commercial RNA isolation kit c and workflow process as previously described. 21 Five hundred microliters of clarified lysate was transferred to a 96-well, deep-well sample plate containing 20 µL of magnetic bead mix (10 µL of lysis/binding enhancer and 10 µL of RNA binding beads), and 300 µL of isopropanol was added. The following plates were loaded onto the automated particle processord,e for nucleic acid purification: sample plate, wash solution 1 plate (300 µL/well), wash solution 2 plate (300 µL/well), and elution buffer plate (90 µL/well). The automated nucleic acid purification procedure consisted of the following steps: lysis/binding for 5 min, one 2-min wash 1, one 2-min wash 2, 1-min dry step, and a 3-min heated elution step.
Each oral swab sample consisted of 4 swabs, collected at 1 time per animal, and suspended in 12 mL of TBTB. a Nucleic acid purification was performed using 150 µL of swab suspension using the reagents and methods described above, except that the 150 µL was added directly into the sample plate containing the 20-µL magnetic bead mix and processed directly as previously described. 21
In parallel, all OF samples were nucleic acid purified using a secondary manual low-throughput nucleic acid purification method. e This method was evaluated in parallel for nucleic acid purification efficacy comparison and for the purpose of confirming the semiautomated method results. For this method, 1 mL of OF was centrifuged at 300 × g for 2 min to remove dirt and debris. Two hundred microliters of clarified sample was used for nucleic acid purification following the manufacturer’s instructions. e XIPC RNA was added to 100 µL of Buffer VXL e at 10,000 copies for each purification.
Multiplex reverse transcription real-time PCR
The mRT-qPCR was performed with ASFV-, CSFV-, FMDV-, and XIPC-specific primers and TaqMan probes (Table 2) using a commercial RT-qPCR kit c according to manufacturer’s instructions. Sequence and final concentration of each oligonucleotide is provided in Table 2. Each 25 µL of mRT-qPCR contained 12.5 µL of 2× multiplex RT-PCR buffer, 2.5 µL of 10× multiplex enzyme mix, 1 µL of 25× primer-probe mix consisting of all oligonucleotides in Table 2, 4 µL of nuclease-free water, and 5 µL of nucleic acid template. Multiplex RT-qPCR was performed using a real-time PCR system. c Cycling conditions (thermal profile) consisted of reverse transcription at 48°C for 15 min (1 cycle), activation and denaturation at 95°C for 10 min (1 cycle), and 40 cycles of amplification at 95°C for 15 sec and 55°C for 45 sec in Fast run mode. Total run time was 75 min. Samples with a quantification cycle (Cq) ≤40 were considered positive, which is consistent with the USDA testing algorithm for ASFV, CSFV, and FMDV.
Data analysis
Polymerase chain reaction efficiency was calculated using the following formula: E = 10(–1/slope) – 1 × 100. R2 was calculated using the RSQ Excel g function with the following description: square of the Pearson product moment correlation coefficient through X and Y data points.
Results
Experimentally infected rope sampling efficacy and presentation of clinical signs
Ropes were introduced to the pigs 2–3 days before experimental infection by throwing knotted ropes onto the pen floor for 10 min to pique the pigs’ interest. Subsequently, the knotted ropes were removed and new unknotted ropes were suspended from the pen wall. This training was performed on 2 consecutive days, and, on the third day, only suspended ropes were provided. Pigs readily chewed the suspended ropes. It was observed that feeding the pigs prior to OF sampling decreased their interest in the rope drastically. After experimental infections, before the appearance of severe clinical signs, pigs readily chewed on the ropes. As the pigs started to show clinical signs, they played less aggressively with the rope, and removal of distractions, such as the presence of technical staff in the room, were required to spur their interest in the ropes. As long as sick pigs were willing to move around, they were willing to at least suck on the rope for a few minutes at a time. At the peak of fever and clinical signs for each disease, infected animals did not move or eat, thus rope sampling was not possible.
ASFV-infected pigs (n = 6 per group, 4 groups total) exhibited fever at 2–4 dpi; clinical signs progressed rapidly following pyrexia. Animals chewed on the ropes every day until the presentation of severe clinical signs, or even sudden death (5–6 dpi, within 48 hr of onset of fever). Clinical signs were observed 1–2 days after onset of fever and consisted of depression, and decreased appetite for Georgia 2007/1–infected pigs, and depression, decreased appetite, diarrhea, and bloody feces for Lisbon 60–infected pigs. CSFV-infected pigs (n = 6 per group, 4 groups total) developed fever at 3–5 dpi, followed by rapid progression of severe clinical disease. Pigs chewed on the ropes every day until 5–6 dpi, the time at which the severity of disease prevented the animals from maintaining their normal activity levels. Clinical signs included depression, decreased appetite, huddling, shivering, diarrhea, and ataxia. FMDV-infected pigs (n = 1 per group, 4 groups total) developed fever at 1–3 dpi and clinical signs at 2–3 dpi; clinical signs consisted of lameness, vesicles on snout and/or feet, blanched coronary bands, and eventual sloughing of hooves over the next few days. Infected pigs chewed on the ropes every day until 2–3 dpi, after onset of several clinical signs. FMD contact pigs (n = 1 per group, 4 groups total) developed fever and clinical signs of lameness, vesicles on snout and/or feet, and blanched coronary bands at 3–7 days postcontact (dpc). Contact animals chewed on ropes until 4–7 dpc when hooves began to slough. FMDV-infected pigs did regain interest in the rope after their temperatures started declining and lesions started healing.
Multiplex RT-qPCR linear dynamic range, relative analytical sensitivity, and efficiency
The mRT-qPCR consisted of previously well-characterized and published assays1,4,21,31 that, at the time of this publication, were in use as single-plex RT-qPCR or qPCR at the USDA-NVSL-FADDL and within the U.S. National Animal Health Laboratory Network (NAHLN). These assays were combined into the multiplex format (Table 2), and the linear dynamic range and relative analytical sensitivity of the mRT-qPCR assay was determined using serial dilutions of pooled purified nucleic acid (purNA) from culture stock viruses. Relative analytical sensitivity in this context refers to the minimum amount of target that can be detected based on serially diluted target inputs and not estimated target copy numbers. Dilutions of viral purNA were preferred over the use of synthetic nucleic acid targets to permit analysis of sensitivity and specificity of target nucleic acid detection in the context of viral genomic nucleic acid and secondary structure motifs such as hairpins. For the relative analytical sensitivity analysis, each purNA pool consisted of purified nucleic acid from ASFV (Kimakia-64, 3.3 × 107 TCID50/mL), CSFV (San Cristobal A tons, 5.2 × 108 TCID50/mL), and FMDV (A Iran 2005, 6.6 × 105 TCID50/mL) thus containing complete pathogen genomic sequences and also mimicking the presence of multiple pathogens in diagnostic samples. Dilutions were performed using pooled purified nucleic acid from OF collected from uninfected control swine to mimic diagnostic samples with potentially interfering host and environmental factors. The pathogen and pooled OF purNA did not contain XIPC RNA, thus each mRT-qPCR was spiked with 500 copies of XIPC RNA. The mRT-qPCR consisted of 4 target detection assays: ASFV, CSFV, FMDV, and XIPC collectively denoted as a 4-plex. Relative analytical sensitivity for each pathogen in the 4-plex was assessed in comparison to the respective single pathogen and XIPC 2-plex (i.e., ASFV and XIPC, CSFV and XIPC, and FMDV and XIPC) and 2 pathogen and XIPC 3-plex (i.e., CSFV-FMDV-XIPC; Fig. 1A–1C). This comparison strategy was performed to assess the multiple oligonucleotides’ combinatory effects on performance.
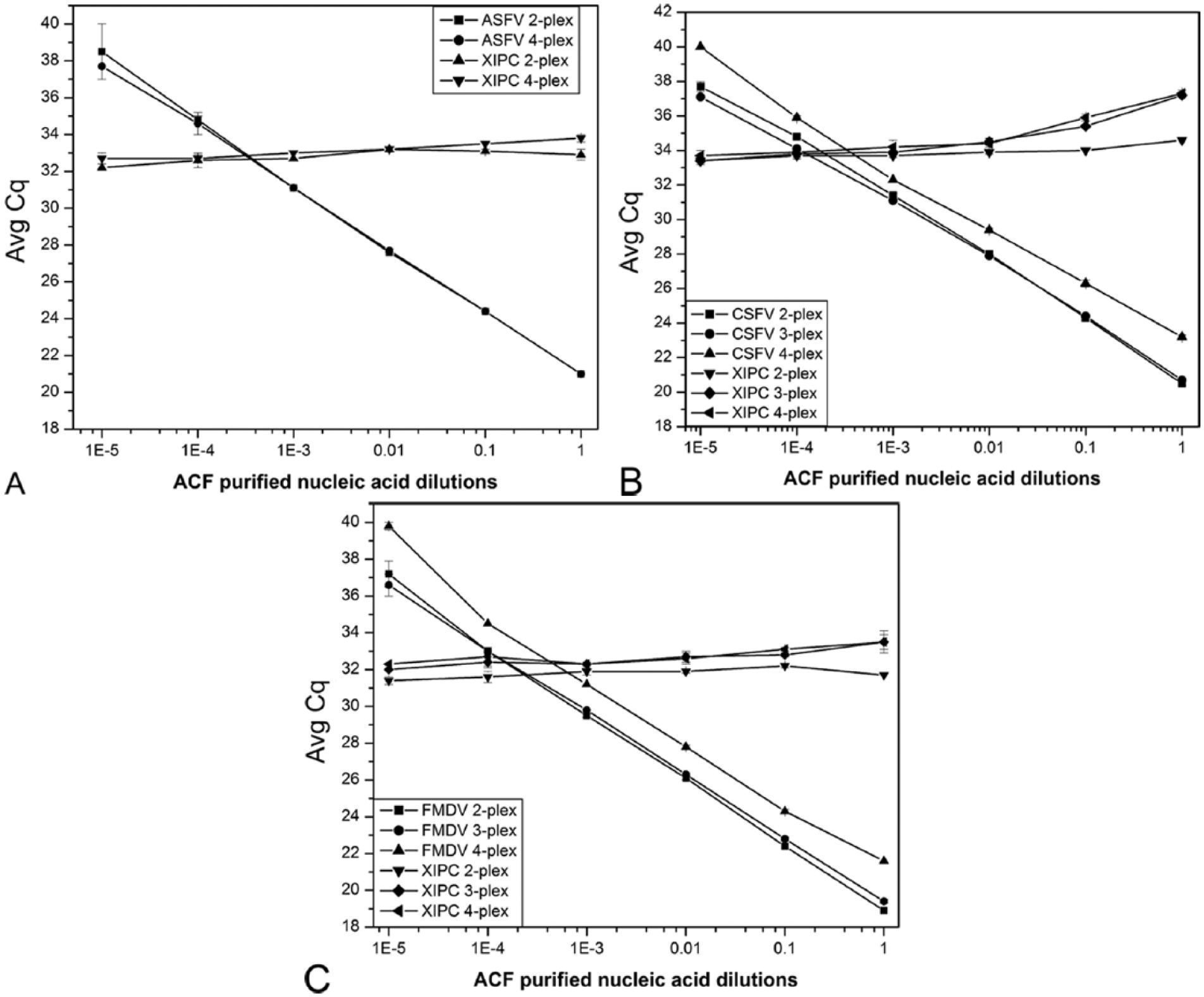
Relative analytical sensitivity of 2-plex, 3-plex (CSFV-FMDV-XIPC), and 4-plex reverse transcription real-time polymerase chain reaction (RT-qPCR) for (
Relative analytical sensitivities of the 4-plex and 2-plex RT-qPCR were comparable for the detection of ASFV as indicated by linear regression plots (Fig. 1A). The relative analytical sensitivity of the CSFV and FMDV 2-plex was comparable to the 3-plex (CSFV-FMDV-XIPC) and higher than the 4-plex as indicated by linear regression plots (Fig. 1B, 1C). Similar results were obtained in 4 other experiments performed on different days (data not shown) to verify the results provided in Figure 1. The 4-plex RT-qPCR exhibited 83.9–98.3% efficiency (R2 > 0.99) for the detection of 10-fold serial dilutions (spanning 5 logs) of each target (Table 3). Copy number equivalents for the limit of detection of the mRT-qPCR were estimated to be <100 copies of each target in a separate mRT-qPCR using linear regression analysis and serial dilutions of an in vitro transcribed CSFV, FMDV, and XIPC RNA, and ASFV DNA (data not shown). In an assessment of specificity, no ASFV, CSFV, or FMDV target amplification was observed using a negative cohort panel of 82 independent swine OF samples kindly provided by collaborators from different geographic regions (i.e., Iowa, South Dakota, Minnesota, North Carolina). Amplification signal was also negative using nucleic acid purified from Bovine viral diarrhea virus 1 and 2, CSFV genome sequence near-neighbors. Since ASFV is the only member of the Asfarviridae family, no near-neighbor is available. FMDV near-neighbors (i.e., Bovine rhinitis A virus and Bovine rhinitis B virus) were not available through vendors or collaborators for evaluation. Because these assays are well characterized, published, and routinely used in the single-plex format at USDA-NVSL-FADDL, analytical specificity testing was limited to near-neighbors based on sequence alignment analysis and virus availability.
Amplification efficiencies for the African swine fever virus (ASFV), Classical swine fever virus (CSFV), and Foot-and-mouth disease virus (FMDV) 2-plex, 3-plex, and 4-plex assays.*
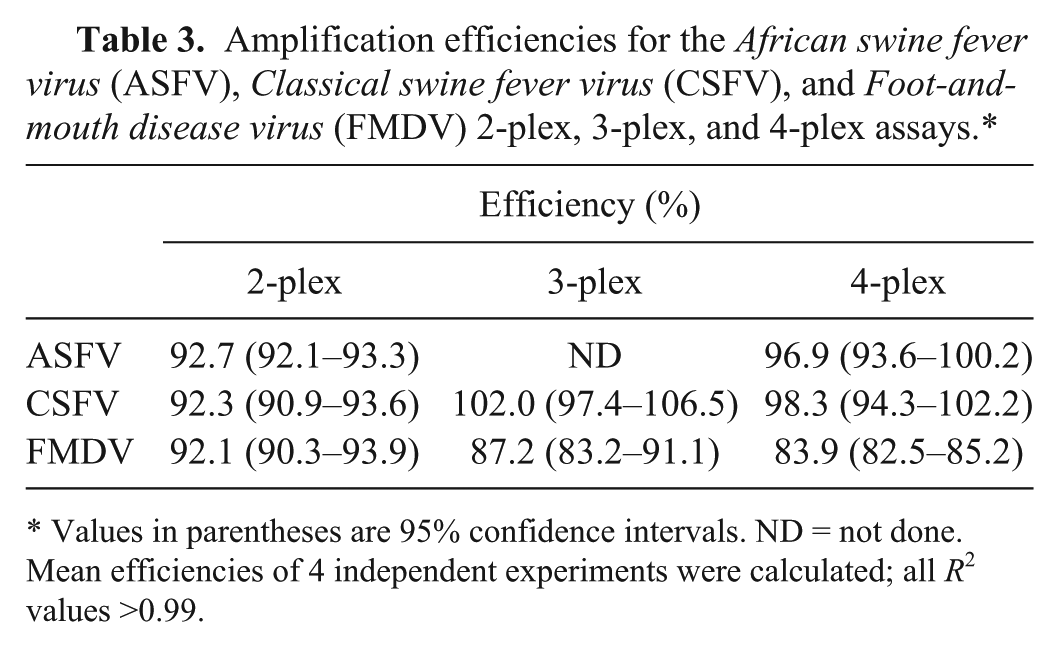
Values in parentheses are 95% confidence intervals. ND = not done. Mean efficiencies of 4 independent experiments were calculated; all R2 values >0.99.
Multiplex RT-qPCR performance evaluation using OF from experimentally infected animals
Performance evaluation of the mRT-qPCR was assessed using OF collected from ASFV, CSFV, and FMDV experimentally infected animals. The XIPC target was introduced in the lysis buffer during nucleic acid purification and was detected in all samples by the 4-plex (Cq range = 31.9–37.9; average = 34.4; standard deviation = 1.4) indicating that nucleic acid purification and PCR performed were valid.
ASFV was detected by 2-plex and 4-plex RT-qPCR with minimal Cq value differences at 3 dpi, prior to the presentation of severe clinical signs in all 4 groups (Table 4); both 2-plex and 4-plex identified 10 of 11 (91%) positive OF samples from infected cohorts. CSFV was detected by the 4-plex RT-qPCR at 5 dpi, coincident with the onset of clinical disease, in 3 of the 4 groups (Table 5). In 5 of the 13 samples tested, the 2-plex and 3-plex RT-qPCR Cq values were lower (1–2 Cq) than the 4-plex RT-qPCR values. The 2-plex identified 10 of 13 (77%) positive OF samples, while the 3-plex and 4-plex identified 8 of 13 (62%) positive OF samples. FMDV was detected in all 15 OF samples by the 2-plex, 3-plex, and 4-plex RT-qPCR (100% positive detection rate). Detection at 1 dpi was observed for all 4 independent FMD study groups, and detection preceded the presentation of clinical signs by 1 day (Table 6). Minimal differences in Cq values were observed among the 3 different RT-qPCR assays; 5 samples exhibited 1 Cq higher value with the 4-plex as compared with the 2-plex RT-qPCR; all other samples exhibited <1 Cq difference. The 2-plex and 3-plex exhibited comparable results for detection of FMDV. Interestingly, the Cq value at 1 dpi for study group 4 was earlier than 5–7 dpi (Cq 22–23 vs. 27–31; Table 6); this may be likely due to higher virus shedding in this study group as both the inoculated and contact animals in this group exhibited clinical signs by 3 dpi. In the other 3 study groups, the contact animals developed clinical signs at 5–7 dpi.
African swine fever virus (ASFV) detection by 2-plex and 4-plex reverse transcription real-time polymerase chain reaction (RT-qPCR).*
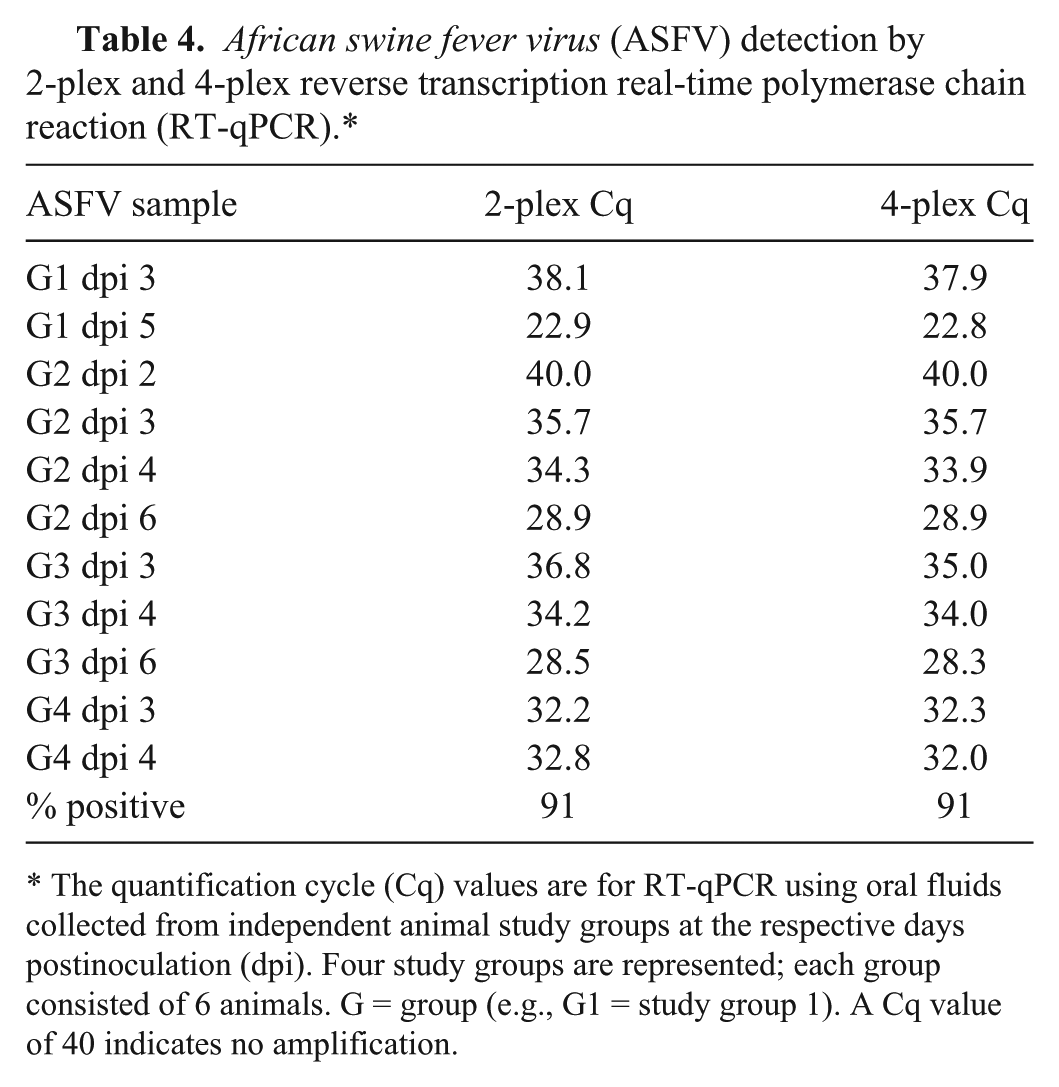
The quantification cycle (Cq) values are for RT-qPCR using oral fluids collected from independent animal study groups at the respective days postinoculation (dpi). Four study groups are represented; each group consisted of 6 animals. G = group (e.g., G1 = study group 1). A Cq value of 40 indicates no amplification.
Classical swine fever virus (CSFV) detection by 2-plex, 3-plex, and 4-plex reverse transcription real-time polymerase chain reaction (RT-qPCR).*
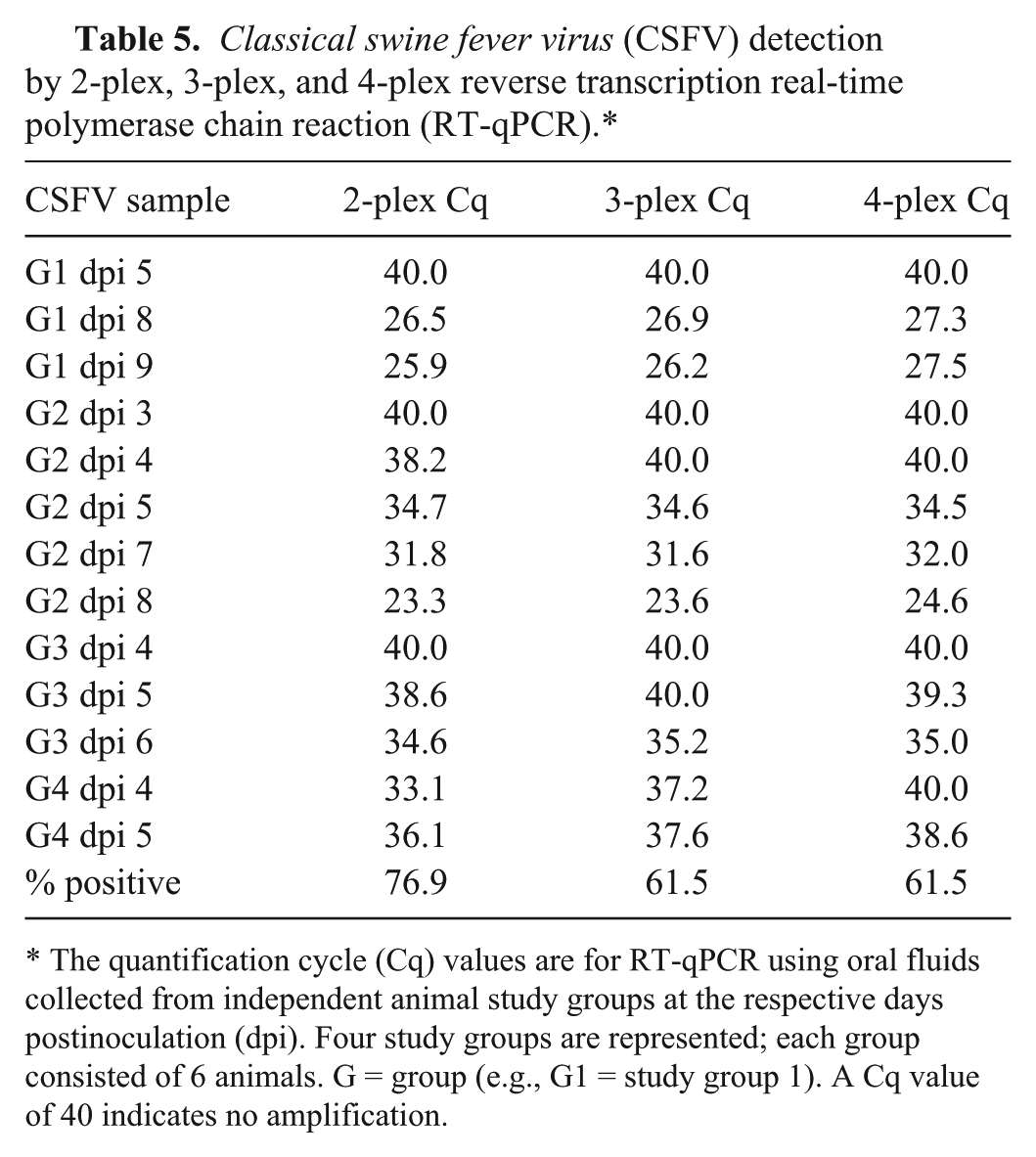
The quantification cycle (Cq) values are for RT-qPCR using oral fluids collected from independent animal study groups at the respective days postinoculation (dpi). Four study groups are represented; each group consisted of 6 animals. G = group (e.g., G1 = study group 1). A Cq value of 40 indicates no amplification.
Foot-and-mouth disease virus (FMDV) detection by 2-plex, 3-plex, and 4-plex reverse transcription real-time polymerase chain reaction (RT-qPCR).*
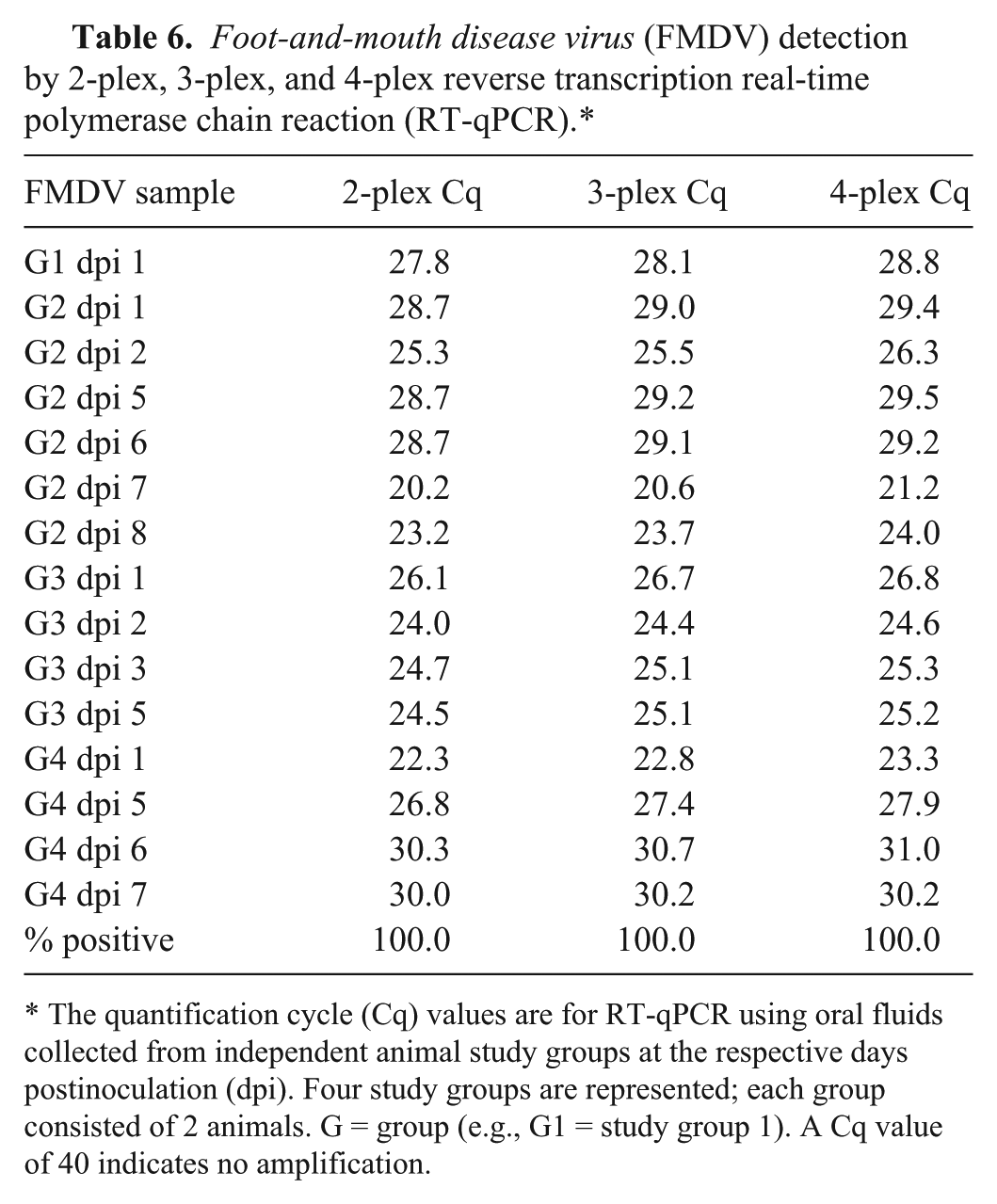
The quantification cycle (Cq) values are for RT-qPCR using oral fluids collected from independent animal study groups at the respective days postinoculation (dpi). Four study groups are represented; each group consisted of 2 animals. G = group (e.g., G1 = study group 1). A Cq value of 40 indicates no amplification.
The observed positive identification rate was lower in CSFV-infected groups (77% by 2-plex and 62% by 4-plex; Table 5). This observation is likely related to lower loads of virus in early dpi samples given the process in which 3 pigs were initially inoculated with the lower virulence Haiti strain at day 0 and the remaining 3 pigs were inoculated at dpi 2 with the higher virulence Brescia strain, which replicates faster to produce higher viral loads.
All samples were also processed using a secondary manual low-throughput, silica filter–based nucleic acid purification method e to confirm the results and to provide an alternative secondary method of nucleic acid purification. The silica filter–based method can be performed manually or automated. Comparable detection results were obtained with this alternative extraction method, adding confidence in the utility of the primary high-throughput nucleic acid purification method (Supplementary Tables 1–3; available at jvdi.sagepub.com/supplemental). For further comparisons, samples were processed in parallel using the USDA-NAHLN RNA and DNA purification methods and tested using the disease-specific RT-qPCR/qPCR assays (data not shown). The NAHLN-employed assays exhibited 36% ASFV (4/11), 54% CSFV (7/13), and 93% FMDV (14/15) positive rates as compared to the 4-plex RT-qPCR, which exhibited 91% ASFV (10/11), 62% CSFV (8/13), and 100% FMDV (15/15) positive rates. Furthermore, NAHLN FMDV assay Cq values were 1–4 Cq higher for 9 samples. The results obtained using the secondary silica filter nucleic acid purification and NAHLN methods provide 2 alternative detection methods to further support the validity of the results obtained with the newly developed multiplex method.
Oral swabs from individual animals in each experimentally infected group were also collected to confirm successful experimental infection and excretion of viruses. Oral swabs from selected individual animals within the respective infected groups were used for nucleic acid purification and detection by the 4-plex RT-qPCR. Successful amplification of the respective pathogen targets was observed, thus providing additional support for the validity of the OF data. The Cq values obtained using oral swabs and OF purified nucleic acid were comparable (Table 7). A comparison of detection sensitivities of OF with the standard detection samples for each agent was beyond the scope of this work but may be appropriate for future validation studies.
African swine fever virus (ASFV), Classical swine fever virus (CSFV), and Foot-and-mouth disease virus (FMDV) detection in oral swabs and oral fluids of experimentally infected animals (study group 2). Oral swabs and oral fluids from a single animal within each infected group were collected and analyzed using the 4-plex reverse transcription real-time polymerase chain reaction to verify presence of viruses in oral cavities.*
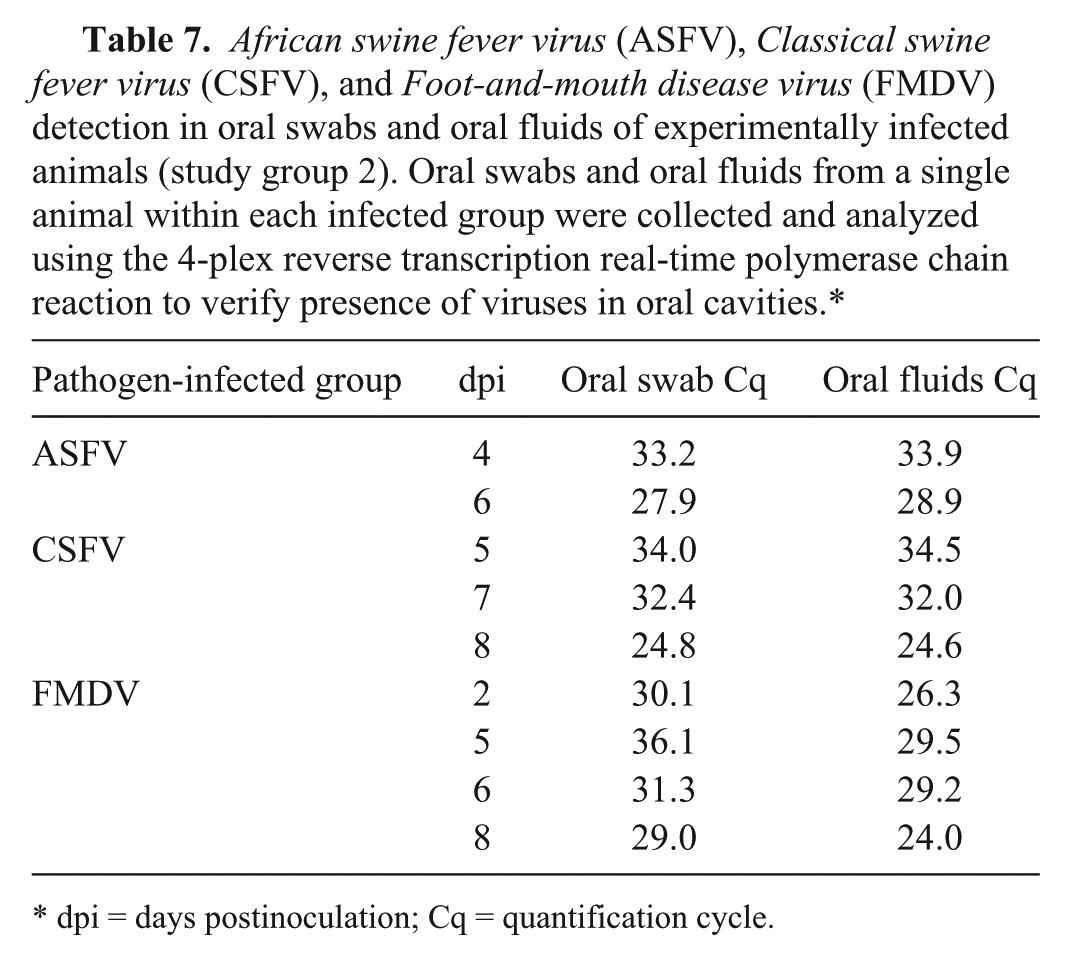
dpi = days postinoculation; Cq = quantification cycle.
Discussion
Early detection is critical to limiting the impact and spread of ASF, CSF, and FMD, and the ability to perform surveillance for all 3 diseases rapidly and cost effectively using a single diagnostic sample and test is highly desirable. Swine in particular, being capable of replicating and shedding FMDV at high titer, have been implicated as an amplifying host in FMD outbreaks,3,10 highlighting the value of swine herd surveillance. Low-virulence CSF represents yet another prominent threat to swine industries with the heightened potential for delayed detection in swine herds. 12 Finally, the spread of ASF in Russia and its emergence in wild boar in Europe, 7 and the emergence of porcine enteric coronaviruses in the United States 23 and other countries have further raised interest in new approaches to swine herd health surveillance.
Although the benefits of OF as a herd sample are now well recognized for several domestic swine diseases, concerns at the outset of this study included 1) the animals’ potential lack of interest in chewing ropes particularly as the severity of clinical disease progressed, and 2) low target detection sensitivity given low pathogen concentrations, poor target stability, and/or copurification of PCR inhibitors. Despite these concerns, the process of OF sampling from pigs was successful for each of the experimental animal studies with ASF, CSF, and FMD. Infected pigs continued to readily chew on ropes before the onset of clinical disease and as long as they could move after onset of fever. Pigs ceased to chew on ropes only after onset of severe clinical signs that restricted movement, but returned to chewing on recovery from illness. Sufficient volumes of OF for testing (5–50 mL) were routinely collected, and the mRT-qPCR displayed a wide dynamic range of nucleic acid target detection. Inclusion of the XIPC internal control further enabled monitoring of the efficacy and validity of the nucleic acid purification and mRT-qPCR. Given these outcomes, rope sampling for OF is anticipated to become more routine for future research studies.
Benefits of the newly developed 4-plex RT-qPCR described herein include feasibility testing in swine OF samples and combined detection and differentiation of ASFV, CSFV, and FMDV in a single-tube reaction. A previous report has described a mRT-qPCR for the simultaneous detection of ASFV, CSFV, FMDV, Suid herpesvirus 1 (pseudorabies; SuHV-1), PRRSV, and PCV-2. 25 Unlike the 4-plex RT-qPCR described herein, the mRT-qPCR from the previous study was not evaluated using OF samples and utilizes the same reporter dye for ASFV, CSFV, FMDV, and SuHV-1 detection, such that positive amplification reported by the dye requires follow-up with 4 single-target RT-qPCR assays for confirmatory diagnosis. 25
The goal of this proof-of-concept study was to assess the feasibility of OF sampling from pigs infected with ASFV, CSFV, or FMDV and to develop a single test for their detection. In keeping with the 3 Rs (replacement, reduction, and refinement) of institutional animal care and use guidelines, this study sought to reduce the number of animals required by leveraging demonstration studies for foreign animal disease infections used to educate veterinary pathologists and diagnosticians. Experimental design and access to animals were therefore limited, resulting in nonconsecutive and nonmatched sampling dates for the 4 independent studies for each disease. In this regard, evaluation of optimal herd size, time to detection, and sensitivity in terms of numbers of infected animals that can be detected in a given herd were also beyond the scope of the study.
Despite these limitations, the study achieved the objectives of feasibility testing of OF sampling and multiplexed detection of ASF, CSF, and FMD, with 4 independent study groups for each disease yielding similar results. ASFV was detected as early as 3 dpi (Table 4), 2–3 days prior to the observance of clinical disease. FMDV was detected as early as 1 dpi, 1 day before the presence of clinical disease (Table 6); and CSFV detection was observed as early as 5 dpi (Table 5), coincident with clinical disease.
Performance of the mRT-qPCR was compared with the respective pathogen 2-plex RT-qPCR and with the USDA-NAHLN protocols. In analytical sensitivity testing, the 4-plex RT-qPCR displayed only a modest reduction in CSFV and FMDV detection sensitivity as compared to the 2-plex RT-qPCR; no loss in sensitivity was noticed for ASFV detection. Use of the optimized OF nucleic acid purification and mRT-qPCR workflow yielded more positive samples than the USDA-NAHLN tests for ASF, CSF, and FMD. Presumably this is because the USDA-NAHLN tests were not optimized for use on OF samples.
Collectively, these findings provide support for future studies to validate the mRT-qPCR and to assess limits of detection in terms of the minimum number of infected pigs for a given disease and herd size. Future field sample validation of such tests, optimized for use with swine OF, should bring closer the potential benefits of cost-effective, safe, and animal welfare friendly swine herd health surveillance for high-consequence transboundary animal diseases.
Footnotes
Acknowledgements
We thank Dr. Tammy Beckham of the Institute for Infectious Animal Diseases for guidance and support. We also thank Drs. Karen Harmon (Iowa State University Veterinary Diagnostic Laboratory), Tracy Otterson (University of Minnesota Veterinary Diagnostic Laboratory), Susanne Hinkley (Neogen Corporation), and Joseph Fent and Perry Harms (Smithfield Premium Genetics) for provision of negative cohort OF samples, and Ping Wu, Fawzi Mohamed, and Elizabeth Clark (USDA-APHIS, PIADC) for their assistance with animal inoculations and the support of the Foreign Animal Disease Diagnostician School. We are also grateful to Jeff Babcock and Michael Santillo, and their staff in the PIADC Animal Resources Branch for expert assistance in animal care, and to Dr. Susanne Hinkley for critical review of the manuscript.
a.
TBTB media, National Veterinary Services Laboratories, Ames, IA.
b.
pBluescript II SK+ plasmid vectors containing DNA sequences specific to ASFV, CSFV, FMDV and XIPC; Blue Heron Biotechnology, Bothell, WA.
c.
MEGAscript T7 high yield transcription kit, MagMAX-96 viral RNA isolation kit, RNA-to-Ct one-step kit manual, MagMax lysis/binding solution concentrate, Path-ID multiplex one-step RT-PCR kit, 7500 Fast real-time PCR system; Applied Biosystems, Carlsbad, CA.
d.
NanoDrop spectrophotometer and KingFisher 96 magnetic particle processor; Thermo Fisher Scientific, Wilmington, DE.
e.
QIAxcel, Tissue lyser, QIAamp cador pathogen mini kit; Qiagen, Valencia, CA.
f.
Primers and probes, Biosearch Technologies, Novato, CA.
g.
Office Professional Plus 2010, Microsoft Excel; Microsoft Corp., Redmond, WA.
Authors’ note
Frederic R. Grau and Megan E. Schroeder contributed equally to this article. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing official policies of the U.S. Department of Homeland Security or the U.S. Department of Agriculture.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This project was funded through an interagency agreement with the Science and Technology Directorate of the U.S. Department of Homeland Security (award HSHQDC-11-X-00528) and by the Institute for Infectious Animal Diseases (formerly, National Center for Foreign Animal and Zoonotic Disease Defense; grant 2010-ST-061-AG0002).