
Abstract
Fungal pathogens threatening the conservation of wildlife are becoming increasingly common. Since 2008, free-ranging snakes across North America have been experiencing a marked increase in the prevalence of snake fungal disease associated with Ophidiomyces ophiodiicola. Diagnosis has historically relied on histology, microbiology, and conventional polymerase chain reaction (PCR). More sensitive methods are needed to adequately characterize the epidemiology. The current study describes the development of a real-time PCR (qPCR) assay for detecting a segment of the internal transcribed spacer 1 region between the 18S and 5.8S ribosomal RNA gene. The assay was able to detect as few as 1.05 × 101 gene copies per reaction. An additional 4 positive cases were detected when comparing a conventional PCR (n = 3) and the qPCR (n = 7) when used on swab samples from 47 eastern massasauga rattlesnakes. The newly developed assay is a sensitive and specific tool for surveillance and monitoring in the conservation of free-ranging snakes.
Studies of wildlife diseases in natural populations aid conservation and recovery of endangered animals. In 2008, 3 eastern massasaugas (Sistrurus catenatus) from the Carlyle Lake (Carlyle, Illinois) population died from a keratinophilic fungal infection with Ophidiomyces ophiodiicola (snake fungal disease [SFD]; formerly Chrysosporium ophiodiicola). 1 Fungal infections in reptiles are typically caused by opportunistic pathogens, infecting animals with a depressed immune system. 6 However, experimental studies show O. ophiodiicola can serve as a primary pathogen and source of mortality in reptiles.1,5 Since 2008, 9 eastern massasaugas from the Carlyle Lake population have been diagnosed with an O. ophiodiicola infection using a conventional polymerase chain reaction (PCR) assay.1,2 In 2011, health assessments of the Carlyle Lake population focused on early detection of the fungus from facial swabs using a conventional PCR targeting a consensus region within the fungal 18S ribosomal RNS (rRNA) gene segment, but none of the 38 animals tested were diagnosed as having O. ophidiicola. 2
The purpose of the current study was to develop and evaluate both a more specific conventional PCR and a real-time PCR (qPCR) assay for characterizing an emerging pathogen, O. ophiodiicola, in snakes. The hypotheses tested in this study were: 1) Ophidiomyces-specific conventional PCR and qPCR assay would be both sensitive and specific for detection of O. ophiodiicola DNA in snakes; and 2) the qPCR assay would detect O. ophiodiicola DNA in more samples than conventional PCR. Having a validated assay is essential when considering the application to characterize the epidemiology of additional free-ranging populations, experimental models, and/or environmental samples that require correct identification.
First, the ChrysoITS-F and ChrysoITS-R primers (Table 1) were designed to target the internal transcriber spacer 1 (ITS1) region that spans between the 18S and 5.8S rRNA with a conventional PCR unique to O. ophiodiicola–based on a National Center for Biotechnology Information (NCBI) search 3 and ITS1 sequence analysis of recent isolates. Each reaction was performed in 25-μL reaction volumes using a commercial kit a with 200 pM of each primer, 0.8 mM of total deoxynucleotide triphosphates, 0.6 U of Taq, and 2 μL of DNA template. The PCR conditions were as follows: denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for 30 sec, annealing at 59°C for 30 sec then 72°C for 30 sec with a final extension at 72°C for 4 min. All PCR products were cut from the gel, purified, and sequenced. The DNA quality and absence of inhibitors were assessed for each sample by demonstrating successful amplification of snake mitochondrial DNA using the 16SAT and 16SBT primers 7 (4 μL each from 10 μM stock; Table 1), 1 commercial lyophilized PCR reagent bead, b and 40 μL of sterile distilled H2O. Following a 3-min denaturation at 94°C, 35 cycles of 94°C for 1 min, annealing at 55°C for 1 min, and 72°C for 1 min were performed, with a final extension at 72°C for 10 min. The DNA quantity per reaction varied from 10–100 ng per reaction.
Primers and probe used for the detection of Ophidiomyces ophiodiiocola.*
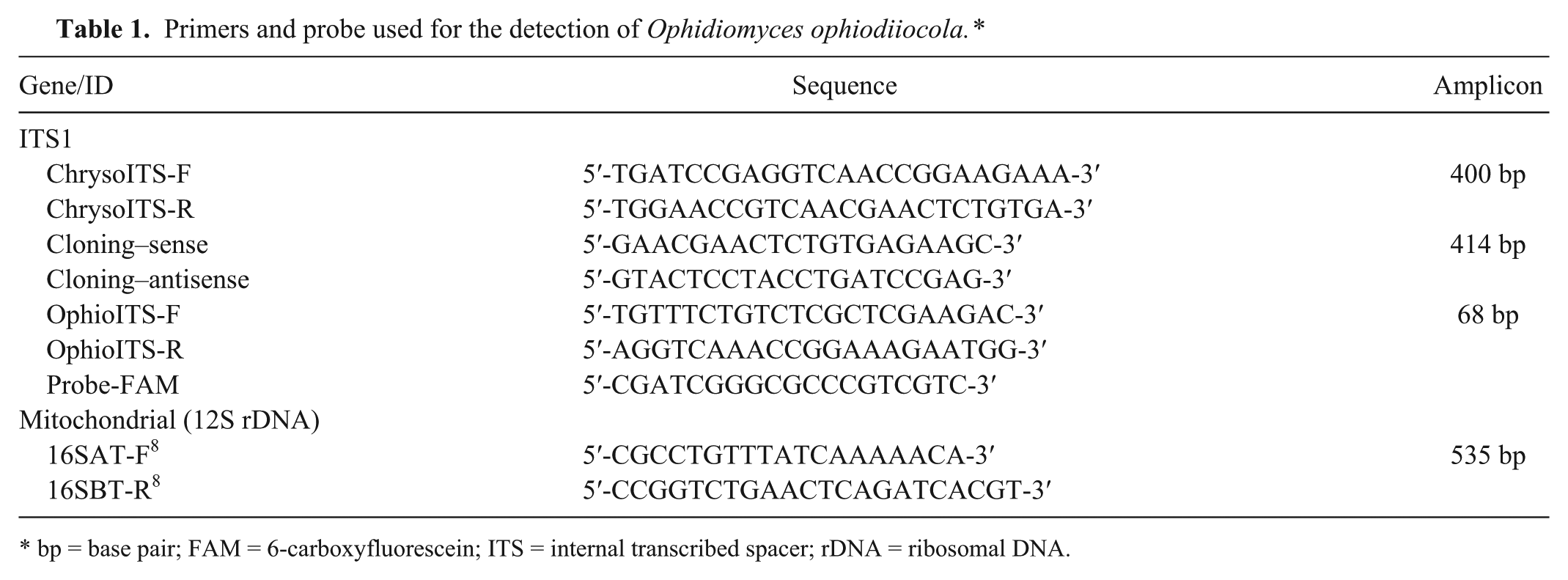
bp = base pair; FAM = 6-carboxyfluorescein; ITS = internal transcribed spacer; rDNA = ribosomal DNA.
Next, a minor groove binder dual-labeled, probe c -based qPCR assay was designed using a commercial software program d to target a 68–base pair (bp) segment of the ITS1 region (Supplementary Fig. 1; available at jvdi.sagepub.com/supplemental). The qPCR assay was performed using the primers OphioITS-F and OphioITS-R and probe Probe-FAM (Table 1). The qPCR assays were performed using a real-time PCR thermocycler, and data was analyzed using the associated software. e Each 25-μL reaction contained 12.5 μL of 2× commercial supermix, f 1.25 μL of the primer-probe (20×), 2.5 μL of O. ophiodiicola target DNA, and water to a final volume of 25 μL. The cycling parameters were as follows: 1 cycle at 50°C for 2 min followed by 95°C for 10 min, then 40 cycles at 95°C for 15 sec and 60°C for 60 sec, and a final cycle of 72°C for 10 min.
A positive control and standard for the quantitation of the qPCR assay was developed by cloning a 414-bp segment of the ITS1 region that spans between the 18S and 5.8S rRNA. This segment was amplified with the cloning primers designed using commercial software d (Table 1) from a template of pure culture of O. ophiodiicola isolated from an infected massasauga. The PCR products were sequenced in both directions at the Keck Biotechnology Center at the University of Illinois (Urbana, Illinois) and compared with sequences in GenBank using Megablast (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The PCR products were then cloned in Escherichia coli using the cloning primers g listed in Table 1. The plasmids were linearized with the restriction enzyme BamH1, h purified, i and quantified using spectrophotometry. Ten-fold serial dilutions of linearized plasmid were made from 2.0 × 102 ng/µL to 2.0 × 10−7 ng/µL. The number of copies of the ITS1 gene segment was calculated using the following formula:
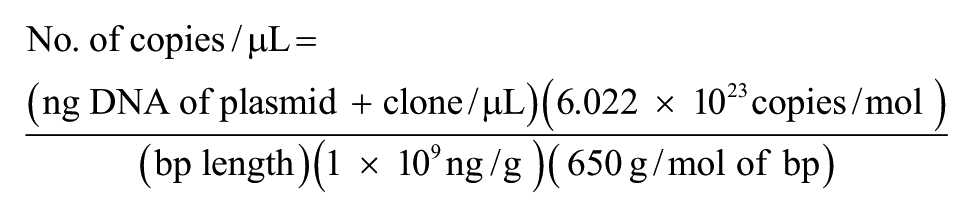
The final number of gene copies of each plasmid dilution ranged from 1.05 × 108 to 1.05 × 101 per reaction. To determine the assay sensitivity, assays were performed in 6 technical repeats of each serial 10-fold plasmid dilution (1.05 × 108–1.05 × 101 gene copies per reaction) within a single run. Standard curves were generated using the threshold cycle (Ct) values of the positive control plasmid dilutions. Inter- and intra-assay variation was determined for both assays by calculating the mean Ct values, standard deviations, and coefficients of variation separately for each control plasmid DNA dilution (replicates) within and between plates, respectively (Table 2). The linear range for the qPCR assay was between 1.05 × 108 and 1.05 × 101 gene copies with an R2 of 0.999 (slope = −3.366). These results indicate high reproducibility between assays at all dilutions. The dynamic range for the qPCR assay was from 1.05 × 108 to 1.05 × 101 gene copies.
Intra- and interassay variability of a real-time polymerase chain reaction assay to detect Ophidiomyces ophiodiiocola DNA.*
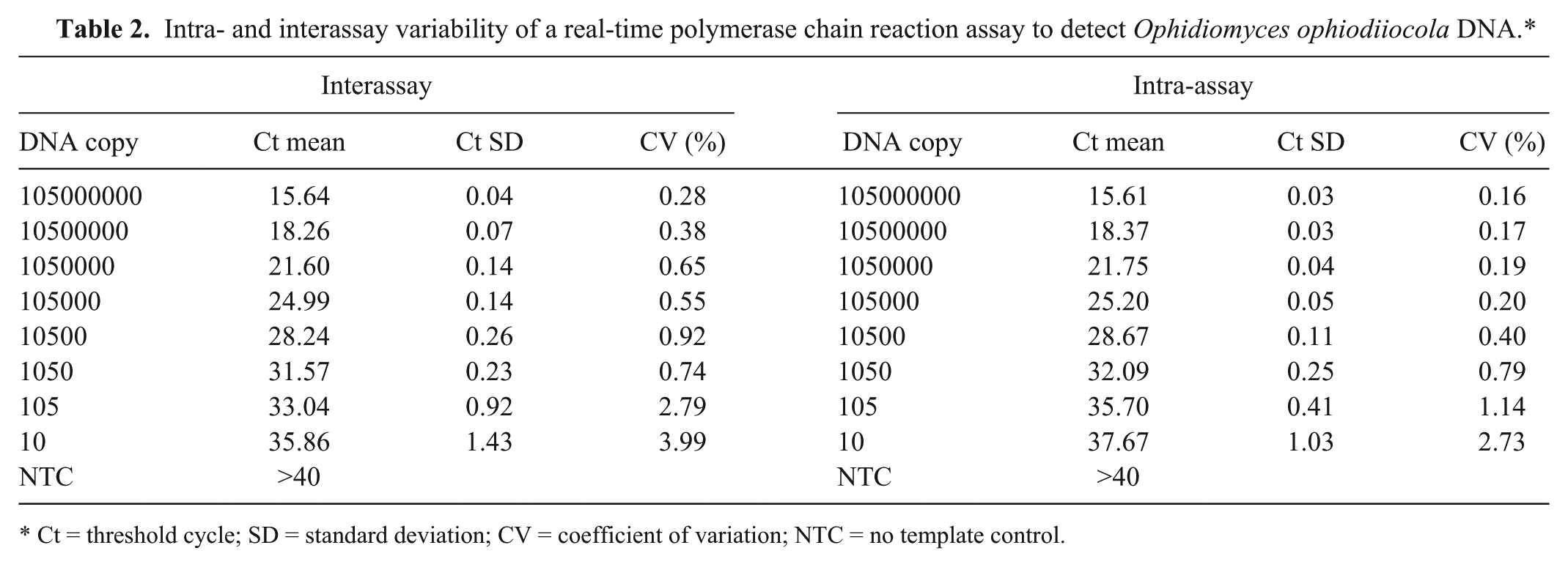
Ct = threshold cycle; SD = standard deviation; CV = coefficient of variation; NTC = no template control.
The specificity of each assay was assessed by demonstrating failure to amplify DNA from common soil fungi as well as fungal organisms recovered from clinical or field isolates that were identified morphologically or by sequencing of the ITS1 region and demonstrating >97% homology to the NCBI database if indicated. The isolates tested included the following: Aspergillus terreus, Aspergillus flavus (FJ487932), Mucor racemosus, Alternaria sp., Blastomyces dermatitidis, Chrysosporium vallenarense (AJ390389), Cladosporium cladosporioides (EU714397), Colletotrichum acutatum (FJ455524), Fusarium lateritium (AF310982), Geomyces sp. (DQ317339), Penicillium verrucosum (KC009829), Pseudogymnoascus destructans, Pseudogymnoascus pannorum (GU062257), and Simplicillium cylindrosporum (AB604006).
To test the utility of the qPCR assay in animal samples, DNA from swabs of 47 free-ranging eastern massasaugas were screened as part of routine demographic and health assessments that were presented to the University of Illinois at Urbana–Champaign Wildlife Epidemiology Laboratory in 2013. A single swab was collected from either the nasolabial pits or from a lesion (when present). The University of Illinois Institutional Animal Use and Care Committee approved all animal use. Nucleic acids were extracted from swabs following 1 hr of incubation in 300 U of lyticase, according to the manufacturer’s instructions. j The quantity and purity of the DNA were evaluated by spectrophotometry. k These DNA extracts, obtained from animals with unknown disease status, were then evaluated using both conventional PCR (ChrysoITS-F and ChrysoITS-R primers) and qPCR (OphioITS-F and OphioITS-R primers). The level of agreement (kappa) was determined between both the qPCR assay and the conventional PCR based on prevalence. The number of DNA target sequences amplified using qPCR were calculated and evaluated for normality using the Shapiro–Wilk test. Mean, median, standard deviation, 95% confidence interval (CI), and 10–90% percentiles were determined for positive cases (number of copies). The prevalence of O. ophiodiicola was determined (categorical variable assigned; 1 = positive, 0 = negative and was based on the Ct value of the lowest dilution of standard curve detected), and binomial confidence intervals were determined for all proportions. All statistical analysis was performed using statistical software.l,m
A total of 47 samples (42 from nasolabial pits or associated lesions and 5 from lesions of the body) yielded 3 (6%; 95% CI: 2–17%) positive snakes using conventional PCR, while the qPCR identified 7 positive snakes (14.9%; 95% CI: 7.4–27.7%), which included all 3 positive by conventional PCR. Products of conventional PCR were sequenced in both directions and confirmed to be 100% homologous to O. ophiodiicola (EU715819). The level of agreement between the 2 assays was moderate (κ = 0.466; 95% CI: 0.04–0.889). The mean fungal DNA quantity in positive snakes was 26,068 copies (95% CI: 2,957–49,181). The mean DNA copy number in samples positive by both methods was 33,833 (95% CI: 0–956,671) and was not significantly different from the samples positive only in the qPCR at 20,245 copies (95% CI: 0–63,250; p = 0.524). Each sample was positive for the presence of reptile mitochondrial DNA (data not shown), confirming that the negative O. ophiodiicola results were not due to poor nucleic acid extraction or inhibition of the PCR.
Multiple copies of gene segments may be present within a single fungal genome, and previous studies have shown that internal controls (i.e., a housekeeping gene) may be necessary when evaluating the rRNA gene segment for quantitation. 4 Those internal controls were not tested in this assay as the objective of the study was to develop a more sensitive assay for detection, rather than absolute quantification. Future studies to determine absolute quantification will need to include these control measures.
This newly developed assay had highly reproducible results with intra- and interassay variability coefficient of variation of less than 5%. The primers were designed to be specific for the ITS1 segment between the 18S and 5.8S rRNA gene, a gene that previous studies have targeted with conventional PCR. 2 While this assay was developed for use in outbreaks involving snakes, the assay has been tested against other fungal organisms and found to be specific.
Ophidiomyces ophiodiicola alone or in concert with environmental pressures are threatening free-ranging snakes in the United States, several species of which are under threat or endangered.5,7,9,10 Despite the endangered status of eastern massasaugas (in the state of Illinois) and the severity of recent disease outbreaks, 1 the epidemiology of this disease is not well understood. Current and future epidemiologic surveys that determine the extent of disease and the range of species affected depend on effective diagnostic assays. The current study has demonstrated that the qPCR assay is reliable, specific, and sensitive for the detection of the ITS1 region between 18S and 5.8S rRNA genes of O. ophiodiicola. The qPCR assay has low variability and is not likely to produce a nonspecific product and therefore is the recommended assay for snake O. ophiodiicola surveys. This assay can be used as a tool in the conservation of snakes by identifying emerging and ongoing outbreaks. This assay is sensitive in detecting as few as 10 copies of the fungal gene segment, thereby increasing utility in identifying early, subclinical, or reservoir disease status detection if those states exist in snakes. As O. ophiodiicola continues to emerge as a major threat to snakes, it is critical that early and accurate identification of epidemics occurs.
Footnotes
a.
Product no. 10342-020, Invitrogen Corp., Carlsbad, CA.
b.
OmniMix HS bead, Cepheid, Sunnyvale, CA.
c.
TaqMan primers, FAM dye labeled; Applied Biosystems, Carlsbad, CA.
d.
Primer Express, Applied Biosystems, Carlsbad, CA.
e.
7500 ABI real-time PCR system, Sequence detection software v2.05; Life Technologies, Grand Island, NY.
f.
TaqMan Platinum PCR supermix-UDG with ROX, Invitrogen Corp., Carlsbad, CA.
g.
TOPO TA cloning kit, Invitrogen Corp., Carlsbad, CA.
h.
BamH1 restriction enzyme, Invitrogen Corp., Carlsbad, CA
i.
QIAfilter plasmid maxi kit, Qiagen Inc., Valencia, CA.
j.
QIAamp DNA mini Kit, QIAamp FFPE tissue kit, BioSprint 96 automated DNA/RNA extraction; Qiagen Inc., Valencia, CA.
k.
EPOCH, BioTek Instruments Inc., Winooski, VT.
l.
SPSS Statistics 22, IBM Corp., Chicago, IL.
m.
MedCalc Software, Ostend, Belgium.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Funding
The author(s) disclosed financial support from the Illinois Department of Natural Resources through the Wildlife Preservation Fund (14-L18W) for this research.