
Abstract
Although advances in nucleic acid sequencing have enabled the discovery of many infectious agents, challenges remain for scientists and veterinary diagnosticians trying to design animal studies with a minimum of variables and to interpret laboratory results. To evaluate pyrosequencing technology as a potential screening method to estimate the virome in pigs, fecal samples were collected from 4 pigs out of a group of 175 that had been raised together since birth. A number of viruses were detected, demonstrating the application of this technology to determine the background “noise” in the pigs. However, pyrosequencing also demonstrated the diversity of viruses within a group of animals and how that can confound experimental design and obscure a definitive diagnosis.
Keywords
The phenomenal advances in nucleic acid sequencing technologies have enabled many discoveries that might not have been made if it were not for this ability to efficiently sequence genomes and process massive amounts of data. Ironically, the technology that has made the concept of deep sequencing and/or metagenomics real may present a challenge for scientists trying to design animal studies having the minimum of variables and for veterinary diagnosticians trying to determine the primary etiologic agent. The assumption that animals procured from a single source were essentially the same was quite acceptable (from the microbiological perspective) in the past, especially in fields of study using outbred large animals. Scientists and diagnosticians investigating swine viral diseases are continually vexed by the discovery of new viruses and how such agents may interact in an individual as well as within a group of animals. Pyrosequencing technology was developed to evaluate a viral metagenomics approach for a potential screening method to determine the viral ecology of pigs and as a potential diagnostic method.
Four pigs were selected from a shipment of 175 animals that were purchased as 3-week-old weaned pigs and delivered to the U.S. Department of Agriculture, Agricultural Research Service, National Animal Disease Center (NADC; Ames, Iowa) for use in separate experiments. Animals were immediately transported from the farrowing house to animal biocontainment facilities as a group in a sanitized trailer dedicated to this purpose. Upon arrival, the pigs were randomly sorted into isolation rooms holding approximately 30 pigs each, and acclimated to the facilities for 1 week. After acclimation, the pigs were randomly assigned to experiments. During acclimation, it was noted that a few pigs from different rooms had developed diarrhea. This was atypical because the source farm historically delivered healthy pigs from a sow herd that was considered of reliable high health status and was routinely negative for Porcine reproductive and respiratory syndrome virus (PRRSV of the Arteriviridae family) and Swine influenza virus. The diarrhea in all but 2 pigs resolved during the acclimation week, and the pigs were assigned to experiments. The more severely diarrheic pigs were housed together in a separate isolation room and, when their condition did not improve, the pigs were humanely euthanized with an intravenous administration of pentobarbital at 6 weeks of age.
In the present study, pigs 1 and 2 were healthy control pigs that did not have any observed diarrhea. Pig 3 was 1 of the 2 pigs chronically affected with diarrhea previously described that was euthanized at 6 weeks of age. Pig 4 had a mild, transient diarrhea following arrival at the NADC that resolved within 1 week at which time it was randomly assigned to an experiment in which it received a PRRSV challenge at 4 weeks of age. Pig 4 had pneumonia at the time of necropsy related to the experimental infection and no overt signs of diarrhea. Approximately 10 ml of feces was collected from the rectum of each pig (6 weeks of age) at the time of euthanasia. Feces were processed to prepare viral nucleic acid libraries for pyrosequencing as previously described. 2 Briefly, fecal samples were diluted in phosphate buffered saline and vortexed. An aliquot of supernatant was collected after centrifugation and filtered through a 0.45-µm filter to remove large eukaryotic and bacterial cell–sized particles. These viral particle–enriched filtrates were then treated with a mixture of DNase and RNase a to digest unprotected or nonencapsidated nucleic acids. Viral nucleic acids were then extracted using standard methods as described. 2 Sequencing was performed using titanium chemistryb,3 and sample barcoding at the NADC. The sequencing reads were binned according to their sample barcodes and then assembled. For pigs 1–4, the total number of sequencing reads per pig was 122,280, 50,665, 50,819, and 179,968, respectively. Assembled contigs and the remaining unassembled reads (singletons) were compared to a database of mammalian viral sequences via BLASTX (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Virus enrichment steps were taken prior to deep sequencing, but as is typical of such studies, nonviral sequences were still detected due to residual host and bacteria nucleic acids. The percentage of nonviral sequences with BLASTX identities to host, phage, bacteria, and unknown were similar to those reported in a prior metagenomic study of pig feces. 2
For comparative purposes, BLAST results were used to categorize the sequences (contigs and singletons) into virus family and genus. The most frequent viral sequence reads were determined for each pig by calculating a ratio of the number of positive reads in relation to the overall total number of reads. This value was normalized to reveal the relative frequency of specific reads from each pig to be compared among the pigs using the following formula: the relative frequency (%) = [(total classified reads for a taxonomic group)/(total number of reads for the animal) × 100]. Positive sequence reads for a taxonomic group were determined by choosing an expectation value (E-value) of ≤10-10 as the cut off for defining BLASTX hits that could be classified based on the best BLASTX high scoring pair. Sequence reads at this cut-off level exhibited highly significant protein sequence similarities with known viruses in the database.
The fecal virome for pigs 1 and 2, the non–virus-challenged control pigs, was similar. Based on normalization of the virus reads, approximately 88% and 34% of the reads for pigs 1 and 2, respectively, matched genus Sapovirus of the Caliciviridae family (Table 1). There was also evidence for additional viruses from other virus families at much lesser frequencies (0.001–0.053% of the reads). In pig 3 (the pig with chronic diarrhea), the most prevalent virus detected was genus Enterovirus of the Picornaviridae family at 2.6% of the reads. In contrast to pigs 1 and 2, the frequency of Sapovirus sequence reads was greatly reduced at 0.008%. Additional virus families were identified at frequencies of 0.002–0.037%. In pig 4 (the pig experimentally infected with PRRSV), the 2 most frequently identified viruses were in the Picornaviridae family with genus Enterovirus at 63% and genus Sapelovirus at 13% followed by other virus families at very low frequencies (0.001–1.706% of the reads) including PRRSV in the Arteriviridae family (.01%). Although PRRSV does induce a systemic disease, there is limited fecal shedding of the virus, which can account for the .01% incidence of genus Arterivirus reads in the fecal sample from pig 4. 4
Normalized number of sequence reads in percentage, and classification of the sequence reads based on protein identity to known viruses.*
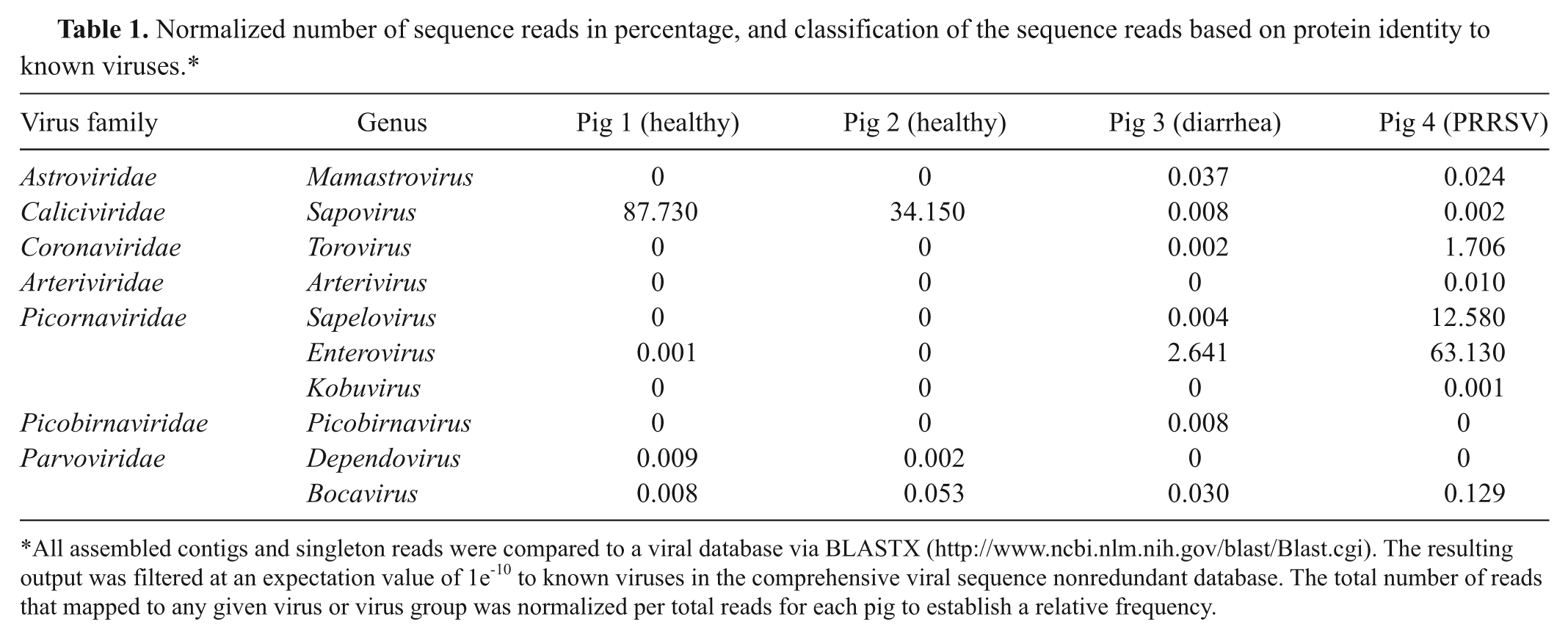
All assembled contigs and singleton reads were compared to a viral database via BLASTX (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The resulting output was filtered at an expectation value of 1e-10 to known viruses in the comprehensive viral sequence nonredundant database. The total number of reads that mapped to any given virus or virus group was normalized per total reads for each pig to establish a relative frequency.
Contigs and singletons that returned a best BLASTX match with E-scores above 1 × 10-10 are sequences that exhibit less similarity to known porcine viruses. Potentially, these sequences may represent more distant “novel” viruses. Further cloning and sequence analysis is required to elucidate the identities of these sequences, and they can serve as input into future experiments designed to discover new viruses not currently in comprehensive databases of viral sequences. There were also reads that had no similarity with any sequences in the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/). Whether there are any highly divergent porcine viruses (unrecognizable by BLAST) among this pool of sequences is unknown.
A variety of viruses were identified in pigs that were healthy or had diarrhea. There were more RNA viruses than DNA viruses detected. The spectrum of viruses observed included the families Astroviridae, Caliciviridae, Coronaviridae, Arteriviridae, Picornaviridae, Picobir naviridae, and Parvoviridae. In general, the findings in the current study are similar to the fecal virome in pigs from a typical commercial swine farm in the United States. 2 In a previous study, 2 Kobuvirus sequences were found at levels 12 times higher in healthy than in sick pigs. In the present study, Sapovirus was identified at a remarkably high frequency in the 2 healthy pigs and barely at all in pigs 3 and 4, the pigs with chronic diarrhea and infected with PRRSV, respectively. Enteroviruses were observed at a much higher relative level in pig 3 (2.6%) and pig 4 (63%) compared to healthy pigs 1 and 2. The findings of apparent fecal shedding of viruses from clinically normal pigs, and that the predominant viruses in a host may differ based on health status, demonstrates the difficulties in establishing the normal background virome for an animal population, even from a single source.
Deep sequencing is a term that has been coined to simply describe the remarkable technology that allows efficient large-scale parallel pyrosequencing of genetic material. The technology enables detection of viruses that might not otherwise be detected by routine diagnostic methods, and such capacity will lead to significant advances in pathogen discovery capacity, full genome sequencing, transcriptomics, metagenomics, and microbial ecology. Clearly, this technology has led to important veterinary discoveries, 1 and certainly will lead to more in the future. The goal of the present study was to establish the deep sequencing and metagenomics analysis methods to characterize the virome of pigs. From this perspective, the trial was successful in identifying a variety of viruses in the feces of pigs. What is interesting is the number of viruses found in the feces of “healthy” pigs, which can create challenges for experimental design and diagnostic interpretations. Traditionally, the assumption has been that pigs raised together would have a common microbial and viral exposure history. This logic would provide the minimum of experimental variables and, thus, simplifies diagnostics. However, it does not account for agents that were not part of the testing process, especially agents unknown to the researcher or diagnostician. Nor does testing at a single time point account for the kinetics of infectious agents within a population.
To date, pyrosequencing technology is cost prohibitive, and there is limited understanding of the presumed interactions of many viruses within the host. Nevertheless, pyrosequencing provides a more comprehensive picture of the viruses coexisting in animals and a greater insight into coinfections, and may lead to new strategies that improve the production of pork.
Footnotes
Acknowledgements
The author thanks Dr. M. Kehrli for critical reading of the manuscript and N. Otis for technical assistance. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture (USDA). The USDA prohibits discrimination in all its programs and activities on the basis of race, color, national origin, age, disability, and where applicable, sex, marital status, familial status, parental status, religion, sexual orientation, genetic information, political beliefs, reprisal, or because all or part of an individual’s income is derived from any public assistance program. (Not all prohibited bases apply to all programs.) Persons with disabilities who require alternative means for communication of program information (Braille, large print, audiotape, etc.) should contact the USDA TARGET Center at (202)720-2600 (voice and TDD). To file a complaint of discrimination, write to USDA, Director, Office of Civil Rights, 1400 Independence Avenue, SW, Washington, DC 20250-9410, or call (800)795-3272 (voice) or (202)720-6382 (TDD). USDA is an equal opportunity provider and employer.
a.
QIAamp MinElute Virus Vacuum kit, Qiagen Inc., Valencia, CA.
b.
Roche–Genome Sequencer FLX system, 454 Life Science, a Roche Company, Branford, CT.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared that they received no financial support for their research and/or authorship of this article.