
Abstract
Infection with Bluetongue virus (BTV) is a significant impediment to the global movement of bovine semen. Repeat testing of blood from donor animals is specified in the World Organization for Animal Health (OIE) Manual for the export of semen from regions where BTV may be present. Screening of blood or semen samples has usually been carried out by virus isolation (VI) either by inoculation of chicken embryos followed by passage onto insect and mammalian cell cultures or in vivo inoculation of sheep followed by serology to detect seroconversion. Direct testing of semen for BTV would enable earlier release of semen samples and avoid repeat testing of the donor, as well as provide an option for releasing batches of semen that were collected without certification of the donor. Quantitative (real-time) reverse transcription polymerase chain reaction (qRT-PCR) assays overcome most of the limitations of other methods and have the potential to provide higher sensitivity. The present study compared 5 qRT-PCR assays, including 2 commercially available kits, for the detection of BTV in semen serially collected from 8 bulls over a period of 90 days after experimental infection. The results of the study show that at least one of the qRT-PCR assays is extremely reproducible and has both very high sensitivity and specificity to reliably detect all available serotypes. The preferred qRT-PCR gave consistently superior results to VI, sheep inoculation, and conventional RT-PCR. Therefore, the assay can be recommended for the screening of bovine semen for freedom from BTV.
Introduction
Reports of the excretion of Bluetongue virus (BTV; family Reoviridae, subfamily Sedoreovirinae, genus Orbivirus) in the semen of bulls have influenced international policy for the movements of both live animals and semen between countries, and sometimes, within countries. In the United States, researchers 2 were able to isolate BTV from the semen of bulls experimentally infected with U.S. serotypes and suggested that this phenomenon may be related to the age of the bull. Subsequently, studies undertaken in Australia to explore these findings in more detail showed that BTV was not detected in the semen of naturally infected bulls, even when these bulls were viremic and that excretion was related to the infection of mature bulls with cell culture–adapted virus. 11 Long after those studies were completed, there was an incursion of BTV serotype 8 (BTV-8) into Europe. 12 It was found that this strain of BTV had unusual biological characteristics, readily crossing the placenta of both sheep and cattle, being shed in the semen of rams and bulls and causing mild disease in cattle.3–5 Such features are unique for a field strain of BTV but are consistent with a cell culture–adapted strain.
Certification of bovine semen for international movement from regions where BTV strains are circulating requires that the donor animals remain seronegative throughout the collection period or that blood is tested for the presence of BTV at the commencement and conclusion of semen collection, and at least every 7 days for virus isolation (VI) or at least every 28 days by reverse transcription polymerase chain reaction (RT-PCR), with negative results.21,22 A direct approach to test semen for BTV would enable earlier release of semen samples and avoid repeated testing of the donor. It would also support retrospective certification of batches of frozen semen that have been collected without concurrent BTV monitoring of the donor.
In earlier studies of experimentally infected bulls 11 by the present authors, a wide range of methods was utilized for the detection of BTV in semen, including the inoculation of cell cultures, chicken embryos, and sheep. The present study compared 5 published real-time reverse transcription PCR (qRT-PCR) assays for capacity to detect a wide range of strains of BTV and then evaluated the performance characteristics of a preferred qRT-PCR assay for its capacity to detect BTV, using the collection of serially collected semen samples from bulls.
Materials and methods
The current project involved a 2-stage process. Initially, 3 published qRT-PCR assays and 2 commercial BTV qRT-PCR kits were evaluated against a representative panel of BTVs, covering all 24 serotypes that had been isolated at that time. Subsequently, a preferred qRT-PCR assay, which had been used in the detection of BTV-25,9,10 was evaluated for its capacity to detect BTV in bovine semen.
Semen samples
Normal semen
Semen was collected from a bull held at a commercial artificial breeding center and was diluted with a commercially available diluent a to sperm concentrations used for routine commercial collections. This extended semen was used for all experiments where semen was spiked with either BTV in tissue culture fluid or blood from naturally infected animals. Additionally, 89 commercially collected semen samples were obtained from individual BTV-free animals and tested to assess the specificity of the qRT-PCR assay.
Spiked semen
To initially compare VI and PCR methods, serial log10 dilutions of Australian reference strains of BTV-1 or BTV-21, 16 which had been passaged in cell culture 3–5 times, were prepared in phosphate buffered gelatin saline (PBGS; pH7.4) and 50 µl mixed with 950 µl of normal extended semen. Two samples were spiked with undiluted cell culture fluid and duplicate samples with dilutions from 10−1 to 10−6, providing a total of 28 samples spiked with BTV from cell culture fluid. In order to measure the impact of semen as a sample on VI and qRT-PCR, the dilutions of BTV-infected cell culture fluid were tested by qRT-PCR and then the theoretical target threshold cycle (Ct) was calculated, assuming that the assay had an efficiency approaching 100%. For this purpose, a 10-fold dilution was assumed to result in an increase in the Ct value of 3.4 units and with an additional 1 Ct for a 2-fold dilution (i.e., a total of 4.4 units for a 1:20 dilution arising from the mixing of virus and semen). Additionally, to test samples spiked with wild-type BTV (not adapted to cell culture), normal extended semen was spiked with blood from animals naturally infected with BTV-1 or BTV-21, the most frequently encountered serotypes in Australia, or in some samples, BTV-7. A total of 60 samples were spiked with BTV-infected blood. This was achieved by preparing a 1:10 dilution of packed red blood cells in sterile, nuclease-free water and then adding 50 µl of the lysed red cell preparation to 950 µl of semen. Nine samples of semen to which PBGS was added and 3 samples spiked with lysed red cells from normal blood were included as negative controls.
Experimentally infected bulls
It was previously shown that BTV was only excreted in the semen of mature bulls when infected with cell culture–adapted virus. 11 Briefly, 8 mature bulls were infected with BTV-1, and semen was collected under commercial conditions twice a week over a period of 4 weeks, weekly to 8 weeks, and at day 91 postinoculation. Semen from these bulls had been retained for long-term storage as both extended semen in straws in liquid nitrogen and as undiluted semen at −80°C. In the original study, to ensure the greatest likelihood of detection of BTV, undiluted semen had been examined in all test methods. Consequently, to facilitate comparison, the undiluted semen samples were used in the current study and were available throughout the period in which infectious virus had been detected, and for 1 collection before and 3 collections after the last detection of infectious virus. A total of 87 samples were available for testing.
Sample identification
The 88 spiked semen samples, 87 semen samples from the experimentally infected bulls, and the 12 negative control samples were arranged in a random sequence. The samples were then given a sequential numerical key to ensure that the identity of individual samples was not known to laboratory staff undertaking the analyses.
Viruses
The capacity of the individual qRT-PCR assays to detect all available serotypes of BTV as well as a range of Australian isolates was compared by testing a collection of reference strains and vaccine seed viruses provided by the OIE Reference Laboratory at the Australian Animal Health Laboratory (Geelong, Victoria, Australia) and a selection of isolates held at Berrimah Research Farm (BRF; Darwin, Northern Territory, Australia), or Elizabeth Macarthur Agricultural Institute (EMAI; Camden, New South Wales, Australia). Blood samples from cattle naturally infected with BTV-1, BTV-7, or BTV-21 were also provided by BRF and EMAI.
Nucleic acid extraction
Prior to extraction, semen samples from the experimentally infected bulls were diluted 1:4 in PBGS, and 50 µl was used for extraction of total nucleic acid. Twenty-five microliters of matching undiluted samples were also extracted. Semen samples that were spiked with BTV from either cell culture or blood were diluted 1:4 in PBGS prior to extraction. All extractions were completed using a commercially available magnetic bead kit b and run in accord with the manufacturer’s instructions. The magnetic beads were handled using an automated magnetic particle handling system. c Purified nucleic acids were eluted in a 50-µl volume and then denatured by heating at 95°C for 5 min. Denatured samples were then either immediately tested or held frozen at −20°C.
Polymerase chain reaction assays and virus isolation
The conventional PCR method published in the OIE Manual of Diagnostic Tests 22 was initially adopted as the benchmark for the current study. During the early stages of the project, it was shown that the method did not have as high a degree of sensitivity as may be needed to detect low levels of virus in semen, and its use was discontinued. Consequently, 3 published qRT-PCR assays, which were considered to have pan-reactivity for the detection of BTV, were evaluated. These included published assays directed at BTV genome segment 1, 14 segment 5, 14 and segment 10.9,10 Two qRT-PCR assays were also available as commercial kits. For ease of description, these assays or kits will be referred to as S1, S5, or S10 for the assays using in-house reagents and K1 and K2 for the commercial kits targeting segment 1 15 and segment 5. 8 The primers and probes for the in-house assembled qRT-PCR assays were manufactured to specification at commercial facilities.d,e Each assay utilized 20 µl of fully formulated master mix, to which 5 µl of template was added. All assays were carried out according to the published protocols except that assays S1, S5, and S10 used a commercial master mix f and were run in accordance with the standard assay conditions specified by the master mix manufacturer. Assays were run for 45 cycles on either a real-time PCR system d or a high-throughput real-time PCR system, d each in standard mode. The background fluorescence was adjusted automatically, and the threshold was set manually at 0.05. Results were expressed as Ct values, being the cycle at which the amplification curve crossed the 0.05 threshold. Threshold cycle values ≥38 were interpreted as inconclusive and ≥40 were considered to be negative. Virus isolation was attempted on spiked semen samples in embryonated chicken eggs (ECE) and cell culture using standard methods. 6
Results
Comparison of assays—virus isolates
The comparative testing of field and vaccine strains of BTV (Table 1) showed that each of the qRT-PCR assays was able to detect the recognized 24 serotypes of BTV. Although there was variability in the sensitivity of each of the assays to detect an individual serotype, overall the S10 assay had consistently higher analytical sensitivity (lower Ct values). These differences were usually greater for the Australian field strains (Table 1) but occasionally there were also poor results for South African strains. Both of the assays that were directed against segment 1 showed poor sensitivity. The commercial kit K1 gave results that would be classified as negative (Ct values >40) for 5 samples. Although the S1 assay gave a positive result with each of the Australian isolates, the sensitivity for the detection of some was reduced to such an extent that it would almost certainly fail to detect them in clinical material. The S5 performed much more consistently across the sample panel but the reduced sensitivity was apparent, with 18 samples giving Ct values that were 3.4–6.6 units (approximately 1–2 log10) greater than the results for the S10 assay. Based on these initial performance criteria, the S10 assay was initially identified as the preferred assay for further detailed evaluation. However, at the end of the study, each assay was used to test a selection of semen samples from the experimentally infected bulls (see below).
Comparison of the performance of quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays for Bluetongue virus (BTV; including commercial kits) for the detection of different field and vaccine strains of BTV.*

Boldfaced entries indicate threshold cycle (Ct) values ≥6 units higher than the S5 assay. Aust = Australia; SA = South Africa; USA = United States of America.
Testing of spiked semen samples
When serial dilutions of BTV were added to semen and tested in parallel by VI and qRT-PCR, it was found that the S10 qRT-PCR had higher sensitivity, detecting BTV RNA in 22 out of 28 spiked samples whereas only 6 of these were detected by VI. The limit of detection of BTV in semen obtained by qRT-PCR was approximately 4 log10 more sensitive than achieved by VI in ECE and cell culture (Table 2), even though VI methods usually examine a volume that is at least ten-fold larger than is examined in the qRT-PCR. The same trends were observed for both BTV-1 and BTV-21. In contrast, testing of the serial dilutions of BTV-infected cell culture fluids that were used to spike the semen samples showed that qRT-PCR was only about 1 log10 more sensitive than VI (data not shown), suggesting that the efficiency of VI was reduced by components of semen.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) and virus isolation (VI) results for semen spiked with dilutions of Bluetongue virus (BTV).
Number positive/number tested.
Estimated threshold cycle value derived from the cell culture fluid, taking all dilution factors into account.
− = negative.
When the qRT-PCR results of spiking of semen with blood are compared with VI in cell culture, the markedly higher sensitivity of the qRT-PCR is again apparent. Of the 60 blood-spiked samples, 42 samples were identified as positive by qRT-PCR. In contrast, virus was only isolated from 9 of these samples, further emphasizing the adverse effects of semen on virus replication in cell culture. Although negative results were obtained for some samples that had been spiked with cell culture–propagated virus or blood from naturally infected animals, these results were not unexpected. For these samples, the material used for the spiking had probably been diluted to a level beyond that where BTV RNA might be consistently detected without testing a large number of replicates. Fifteen of the 17 samples that had been spiked with blood had “predicted” Ct values >35 and the other 2 samples had Ct values of 34. Further, while these values assume an assay efficiency of 100%, this does not take into consideration any inhibitory effects that components of the semen or blood might have.
After the dilution factors from adding a very small amount of blood or cell culture–propagated virus to the semen are taken into account, there is limited evidence of inhibitory effects on the PCR for most samples. When the actual and “predicted” Ct values are compared, only 4 out of 62 samples show a difference of >2 Ct units, with 3 samples showing differences of 3 Ct and 1 sample showing 4 Ct units (data not shown).
Although the preferred assay has high sensitivity, it does not lack specificity. Negative results were obtained for the 12 negative control samples that were randomly included in the test sample panel and for each of the 89 individual semen samples from bulls that had not been infected with BTV.
Samples from BTV-infected bulls
All “raw” semen samples were tested both undiluted and at a 1:4 dilution. Three samples of undiluted semen gave a positive PCR result, but BTV was not detected when these samples were diluted 1:4. For the remaining samples, in which BTV was detected in both the undiluted and diluted semen, the results of the undiluted samples gave a mean Ct value that was approximately 2.5 units lower. The results subsequently used in any comparisons are for the undiluted semen.
The virus detection results listed in Table 3 consist of a composite of the VI results and virus detection by sheep inoculation. In the original project, 13 it had been shown that in vivo assay by the inoculation of sheep provided the highest sensitivity, firstly because a larger volume (2 ml) of sample could be examined and secondly because toxic effects in VI systems (embryonated eggs or cell culture) could be avoided or minimized. The increased sensitivity of the qRT-PCR over other methods is clearly apparent (Table 3; Fig. 1). The qRT-PCR was consistently superior to the virus isolation and/or virus detection methods, which failed to detect BTV in some individual samples in a sequence of positive samples from the same bull (bulls F and G). Additionally, BTV RNA was consistently detected in semen samples for a longer period in each animal in which BTV was detected in semen (bulls A, B, F, G), with 3 giving positive results 60 days after the last detection of infectious virus (Fig. 1). In contrast, when BTV had not been detected at all in the semen of a bull by sheep inoculation (bulls C–E), negative results were also obtained in the qRT-PCR. Erratic results were obtained for bull H. Although BTV had been detected in 2 semen samples from this bull when first screened by sheep inoculation, repeat attempts to isolate or detect virus were unsuccessful. Two positive semen samples were detected, by qRT-PCR, 6 and 20 days postinoculation. One of these positive results was for a collection that had given suspect results by sheep inoculation. The data clearly show the superior sensitivity of the qRT-PCR for the detection of BTV in semen, despite the larger volume of sample that was used for VI in cell culture or screening by sheep inoculation.
Virus isolation (VI) and quantitative reverse transcription polymerase chain reaction (qRT-PCR) results on semen from experimentally infected bulls.
Includes virus detection by isolation in embryonated chicken eggs, cell culture, and sheep inoculation.
No virus detected.
Virus detected.
Virus initially detected but could not be confirmed.
Not available for testing.
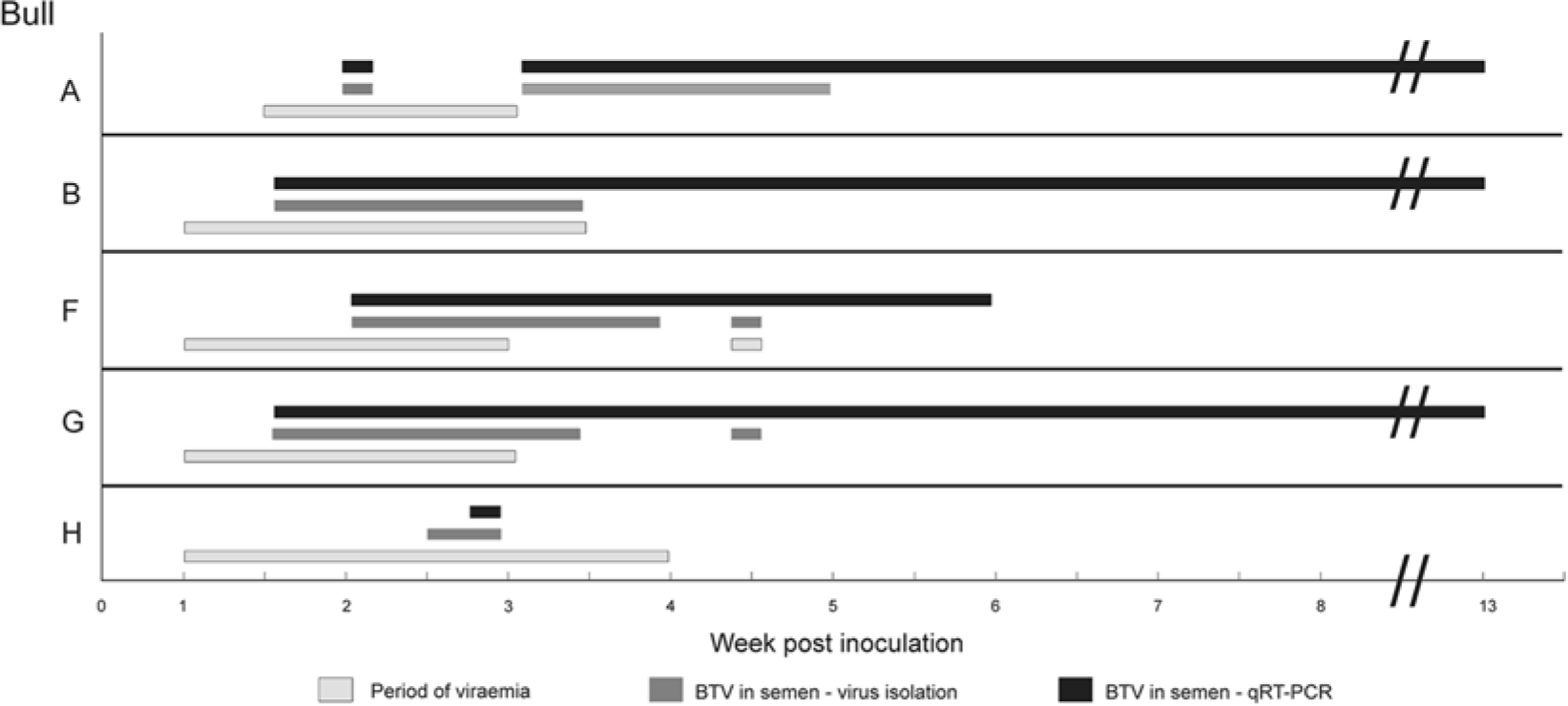
Duration of viremia and virus detection in semen of mature bulls infected with laboratory adapted Bluetongue virus serotype 1 (BTV-1). qRT-PCR = quantitative reverse transcription polymerase chain reaction.
Finally, when a random selection of these nucleic acid extracts were tested in each of the assays under study (Table 4), the superior performance of the S10 assay is again apparent, but not as marked as when testing a wider range of viruses. The 2 assays (S1 and K1) directed at sequences from segment 1 gave identical sensitivity but the commercial kit had very poor specificity, producing a large number of false-positive or nonspecific reactions as observed by the amplification profiles.
Comparison of the performance of quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays (including commercial kits) for the detection of Bluetongue virus (BTV) in semen from experimentally infected bulls.

Denotes the number of samples that were tested in this assay that gave negative results.
Number of samples giving positive results.
Does not include 35 samples that gave false-positive results.
Discussion
Overall, the results of the current study indicate that the preferred qRT-PCR, based on detection of a sequence from segment 10 of the BTV genome, is both sensitive and specific and is suitable for the direct detection of BTV in semen samples. Other testing has shown that this assay also has very high sensitivity and specificity when testing blood samples. The assay maintains good sensitivity for the detection of BTV RNA from all 26 known serotypes1,9,10 and a wide range of genotypes. While some of the other published assays perform well with samples containing certain serotypes, unfortunately they do not have a level of performance comparable to the S10 assay over all serotypes. A similar observation made by Swiss researchers when BTV-25 was first recognized lead to the development of this S10 assay.9,10 The results for the S5 assay, based on the same primers and probe used in the assay optimized for the screening of semen from bulls infected with BTV-8,18,19 provides a good example of this variability. Even though it was the second best assay and detected many isolates with similar sensitivity to the S10 assay, there were instances where differences up to 6.6 Ct (almost 2 log10) were observed. Such differences may be critical when testing clinical samples that have moderate to low levels of BTV RNA present.
While the results show the marked improvement in sensitivity when compared with VI in cell culture, they should be interpreted with caution. The results do not necessarily mean that cell culture systems lack sensitivity because cell culture is only detecting infectious whole virus whereas the PCR assays are detecting total viral RNA that can be available from degraded and noninfectious particles. In many instances BTV RNA was detected on several occasions after infectious virus was detected. Belgian researchers 18 have described similar results when testing semen of bulls naturally infected with BTV-8. Attempts to isolate BTV-8 from 9 out of 48 PCR-positive samples were successful in only 4 samples with the highest viral load. While these differences between VI and qRT-PCR may appear to be a concern, for importing countries this provides added security that infectious samples are unlikely to be imported.
There are several important considerations when comparing these assays. In general, factors favor the “traditional” diagnostic methods. First, VI was carried out when the samples were fresh and had not been frozen, or had recently been frozen in liquid nitrogen. The current qRT-PCR evaluation was undertaken more than 15 years later. The second, and perhaps the most important consideration, is that the original virus detection results that are included in Table 3 represent the most successful result for these samples after VI in cell cultures and ECEs and by sheep inoculation where the latter employed a sample volume of 2 ml. The use of a markedly larger sample volume alone (e.g., approximately 400 times for sheep inoculation) should provide higher sensitivity in samples that have a low virus titer, as many semen samples do. However, testing a large sample volume in an assay presents a considerable logistical challenge that leads to a higher cost, as well as the need to exploit a large amount of an often valuable sample. The extremely high sensitivity of the qRT-PCR overcomes these problems. In order to maximize the detection of low levels of virus and perhaps to compensate for nonuniform distribution of virus in a collection, testing of multiple straws of extended semen has been recommended.18,23 Whether the higher sensitivity of the S10 assay will avert the need to test semen from multiple straws is yet to be proven but it may be possible to pool the semen from a number of straws for testing.
The capacity of the qRT-PCR assay to detect residual RNA from noninfectious virus may overrepresent the number of samples that still contain infective BTV. In 3 of the 4 bulls where there was prolonged excretion of virus in semen, BTV RNA was still detected at day 91 at the end of the study, more than 60 days after the last detection of infectious virus. In the other bull, BTV RNA was detected for 40 days longer than infectious virus. The studies of BTV-8–contaminated semen also found BTV RNA for several months after animals were infected. 18 These results are not surprising as BTV RNA has been detected in cattle blood by conventional PCR for prolonged periods 12 and, in a previous study of 10 cattle infected with 2 different Australian serotypes (1 and 23), the S10 assay consistently detected very low levels of BTV RNA in blood for 7 months from the time of inoculation (X. Gu, P. D. Kirkland, and P. Hick, unpublished observations). Nevertheless, negative results were eventually obtained.
While there have been concerns that bovine semen contains inhibitors of PCR, it is apparent from the results of the current study that components of bovine semen adversely affect the sensitivity of BTV isolation in cell cultures, most likely on the replication of BTV, perhaps as a result of cytotoxicity or by affecting entry of virus into cells. This is reflected in the differences in virus titers detected in BTV-infected cell culture fluids before and after addition to semen. Negative qRT-PCR results were obtained for some samples that had been spiked with either cell culture–propagated virus or blood from naturally infected animals. However, the results were expected as the material used for the spiking had been effectively diluted beyond the level that would consistently allow the detection of BTV RNA in a single assay that uses a small volume for both extraction and PCR. Furthermore, this does not take into consideration any inhibitory effects that components of the semen might have. When the dilution factors from adding a very small amount of blood or cell culture–propagated virus to the semen are taken into account, there is little evidence of inhibitory effects on the PCR in most samples (see Table 2, comparison of “predicted” and observed Ct values). The use of a magnetic bead–based nucleic acid extraction system, combined with a semiautomatic particle handling system, is believed to have given a preparation that has a higher level of purity and with fewer inhibitors than was previously possible when testing undiluted semen. Although the quantity of BTV RNA detected appeared lower than predicted in some spiked samples, BTV RNA was readily detected and at much higher levels in the semen of the experimentally infected bulls, and more readily than by conventional VI methods. The current authors have also obtained similar results for the detection of Bovine herpesvirus 1 and Bovine viral diarrhea virus in undiluted bovine semen as well as Bungowannah virus in undiluted porcine semen (R. Davis, X Gu, D. Finlaison, and P. D. Kirkland, unpublished data).
Although the adverse effects of semen on qRT-PCR appear to have been minimized, the inclusion of appropriate “internal” and “exogenous” controls to monitor the efficiency of the nucleic acid extraction and amplification is recommended.7,20 The addition of an “exogenous” target to the sample may be preferred over the detection of host genomic targets because the “external control” target can be included at a suitably low level to readily detect the presence of modest levels of inhibition. In a preliminary study to identify an appropriate exogenous control, a commercially available construct g was used to reexamine some of the semen samples that were still available. All samples from bull B gave comparable results in the BTV qRT-PCR, and the exogenous control was detected at the expected level (M. Frost and P. D. Kirkland, unpublished observations).
While the preferred real-time PCR assay has very high diagnostic and analytical sensitivity, it appears that this has not come at the expense of specificity with all BTV-free samples giving negative results. The assay has also been used extensively for the detection of BTV in blood samples with similar success by the current authors. Similarly, there was no evidence of cross-reactivity with a range of other viruses likely to be detected in cattle, even the most closely related orbiviruses from the Epizootic hemorrhagic disease virus serogroup. In conclusion, the present results would indicate that the S10 assay is well suited for testing of cattle semen to detect BTV or to demonstrate an absence of recent exposure to BTV strains by testing of blood samples.
Footnotes
Acknowledgements
Technical assistance in Darwin was provided by Margaret Harmsen. The technical assistance of the staff of the Virology Laboratory at EMAI, Camden, is appreciated.
a.
Bioxcell, IMV Technologies, L’Aigle, France.
b.
MagMax 96 viral RNA isolation kit, Ambion, Austin, TX.
c.
Kingfisher 96, Thermo, Vantaa, Finland.
d.
7500 Fast Real-Time PCR System, 7900HT Fast Real-Time PCR System; Applied Biosystems, Foster City, CA.
e.
Biosearch Technologies, Novato, CA.
f.
Ag Path-ID one-step RT-PCR kit (AM1005), Ambion, Austin, TX.
g.
Xeno RNA control, Ambion, Austin, TX.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this work was provided by Biosecurity Australia, Canberra, ACT, Australia.