
Abstract
Equine arteritis virus (EAV) causes contagious equine viral arteritis, characterized by fever, anorexia, conjunctivitis, nasal discharge, dependent edema, abortion, infrequent death in foals, and establishment of the carrier state in stallions. The World Organization for Animal Health (OIE) defines a horse as seropositive if the serum neutralization (SN) antibody titer is ≥1:4 to EAV. However, determining the SN titer is time-consuming and requires specific laboratory facilities, equipment, and technical expertise to perform. Furthermore, interpretation of the SN titer of some sera can be difficult because of nonspecific cellular cytotoxicity of particular samples. Finally, the problem of interlaboratory variation also exists with SN assays. For these reasons, an alternative serologic test is desirable; however, none of the reported tests have equivalent sensitivity and specificity to the SN to be generally adopted. In an attempt to improve on a previously developed competitive enzyme-linked immunosorbent assay (cELISA) using EAV gp5-specific neutralizing monoclonal antibody (mAb) 4B2, the current study developed a modified protocol substituting the non-neutralizing mAb 17B7 for the neutralizing mAb 4B2; this along with several modifications of the test procedure improved the performance of the test. The relative specificity of the revamped cELISA was 99.8% when evaluated with 2,223 SN-negative sera. The relative sensitivity was 95.5% when evaluated with 246 SN-positive sera. This new cELISA was not affected by the presence of non–EAV-specific cytotoxicity in sera as observed in the SN assay. The results indicate that this new cELISA may be a viable alternative to the SN assay and merit additional validation.
Introduction
Equine arteritis virus (EAV; order Nidovirales, family Arteriviridae, genus Arterivirus) is the causative agent of equine viral arteritis. 2 The virus may cause all or a combination of clinical signs including fever, anorexia, conjunctivitis, nasal discharge, dependent edema, abortion, and infrequently death in young foals. 24 This disease is mainly transmitted by respiratory and venereal routes from infected to uninfected horses. An additional serious consequence of infection is that 10–70% of infected stallions become clinically inapparent carriers following recovery from the acute phase of the infection. 23 Carrier stallions are a significant source of infection for mares in that the stallions shed virus constantly in semen. The stallions can transmit EAV by natural service or artificial insemination. Therefore, careful screening and early identification of carrier stallions using sensitive and specific diagnostic tests are critical to the effective control of EAV on breeding farms.
Equine arteritis virus has a positive-sense RNA genome. It is closely related to Porcine reproductive and respiratory syndrome virus, Lactate dehydrogenase-elevating virus of mice, and Simian hemorrhagic fever virus.3,8,12,16 Equine arteritis virus has a genome of approximately 12.7 kb with 10 open reading frames (ORFs). Replicase polyproteins 1a and 1ab (from ORF1a and ORF1b) are cleaved into at least 13 nonstructural proteins. The structural proteins include E (ORF2a), gp2b (ORF2b), gp3 (ORF3), gp4 (ORF4), gp5 (ORF5), ORF5a protein (ORF5a), M (ORF6), and N (ORF7).11,20 The gp5 protein carries the major neutralizing antibody epitopes and is the most reactive structural protein in binding with antibody in EAV-positive sera.1,5
Various assays have been developed to detect EAV antibody, and some of those assays have been used in the field. However, except for the serum neutralization (SN) test, most of these assays did not reliably identify EAV-seropositive horses. In addition to false-positive reactions due to previous exposure to non-EAV biologicals reported using indirect ELISA methods,4,7 most other ELISA assays lack optimal sensitivity and/or specificity relative to the SN assay. The SN assay principally detects antibodies to immunodominant gp5, and is considered the most sensitive assay to detect EAV-specific antibodies in horse serum. The SN assay is, to date, the prescribed test for international trade by the World Organization for Animal Health (OIE); a SN antibody titer of ≥1:4 is considered positive for EAV. However, the SN assay is time-consuming and requires specific laboratory facilities, equipment, and technical expertise to perform. Considerable interlaboratory variation in results is another problem not infrequently encountered using this assay,17,19 and difficulty in interpretation of SN results due to nonspecific cellular cytotoxicity in sera from horses vaccinated with a particular commercial herpesvirus vaccine in Europe has been reported. 17
A previously developed competitive enzyme-linked immunosorbent assay (cELISA) that was termed a blocking ELISA was based on an epitope on EAV gp5; reported sensitivity and specificity were 99.4% and 97.7%, respectively. 6 This cELISA is not adversely affected by previous exposure of horses to non-EAV biologicals. However, interpretation of the assay was complicated by the fact that 12% of the sera evaluated were classified into a suspect range. If the suspect range is considered positive, then the specificity is lower than the reported 97.7%. Whereas, if the suspect range is considered negative, then the sensitivity is lower than the reported 99.4%. 6 Furthermore, some SN titered sera (≥1:32 if undiluted) were negative in this assay (prozone phenomenon). 6 The current study describes improvement of a gp5-based cELISA as part of a continuing effort to develop simpler and more reliable serological assays for detection of EAV.
Materials and methods
Horse sera tested
An antibody-negative reference serum was prepared by screening sera from uninfected and nonvaccinated horses that were negative for neutralizing antibodies to EAV by the SN assay. A positive reference serum, which had an SN antibody titer of >1:512, was obtained from a horse vaccinated with a modified live EAV vaccine. a Nineteen well-characterized sera from horses with natural infection (S1–4, S10–13, S18, and S19) or vaccinated with a modified live EAV vaccine (S5–9, S14–17) were included as a reference panel of positive sera. Virological and serological status was monitored by polymerase chain reaction and SN test before serum collection. A total of 2,450 field serum samples that had been submitted for SN testing in the United States and Canada were also included in the current study. Seventy horse sera, 31 EAV negative by SN assay and 39 EAV positive by SN assay, all with non–EAV-specific cytotoxicity titers ranging from 1:4 to 1:32 in the current OIE-prescribed SN assay, were included to determine if cytotoxicity would interfere with the diagnostic performance of the new cELISA.
Mouse monoclonal antibody against EAV
Production and characterization of mouse monoclonal antibody (mAb) 17B7 (immunoglobulin G1 isotype) has previously been described. 9 The specificity of mAb 17B7 is to the glycoprotein gp5 of EAV.
Propagation and partial purification of EAV
The Bucyrus strain of EAV in 5 or lower passage was used to develop the antigen for the cELISA. The EAV seed used tested negative for contaminations by Bovine viral diarrhea virus and major animal origin viruses in 9CFR (Code of Federal Regulations) testing performed at the VMRD R&D laboratory (Pullman, WA) as well as a confirmatory testing performed at the U.S. Department of Agriculture Center for Veterinary Biologics laboratory (Ames, IA). The virus was propagated in primary equine spleen cells that were free of adventitious viruses. Confluent cell monolayers in 850-cm2 roller bottle flasks were inoculated with a multiplicity of infection of approximately 0.0005. After incubation for 2 or 3 days, when virus-induced cytopathic effect reached a maximum, cells and culture supernatant were combined and centrifuged at 4,000 × g for 10 min at 4°C to eliminate cell debris. The procedure for partial purification of EAV antigen was according to a previously described method with some modifications. 6 Briefly, the virus supernatant was centrifuged b at 21,000 × g for 18 hr at 4°C. The supernatant was discarded, and the virus-containing pellet was resuspended to 1/50 of its original volume in phosphate buffered saline (PBS) and centrifuged at 4,000 × g for 10 min at 4°C. The resulting supernatant (S1) was saved. The pellet was again resuspended in PBS to 1/50 of its original volume and centrifuged at 4,000 × g for 10 min at 4°C. The resulting supernatant (S2) and S1 were combined and subjected to ultracentrifugation at 45,000 × g for 4 hr at 4°C. After centrifugation, the supernatant was discarded, and the virus-containing pellet was resuspended in 0.85% saline to 1/1,000 of its original volume. Triton X-100 c was added to make 0.2% final concentration, the mixture stirred overnight at 4°C, and then held at 37°C for 1 hr. The solubilized preparation was centrifuged at 15,000 × g for 10 min at 4°C, and the resultant supernatant contained the antigen used in the cELISA.
Competitive ELISA and serum neutralization assay
A working dilution of EAV antigen that gave an optical density (OD) reading of approximately 0.8 at 450 nm (A450) when tested against the negative reference serum was made in 0.05 M carbonate buffer (pH 9.6). The working dilution was determined by coating 96-well plates
d
with 2-fold dilutions of the antigen using 50 µl/well. The plates were sealed and held 2 hr at 37°C, and then blocked with 200 µl/well of blocking buffer.
e
The antigen-coated plates were dried overnight at 25°C, and stored individually in
The stored plates and all other reagents for the cELISA procedure were warmed to room temperature. First, 50 µl/well each of reference-positive and -negative sera and undiluted test sera were added to the antigen-coated plates. The plates were incubated for 2 hr in the 17B7-based assay before washing. Then, an optimized dilution of mAb 17B7 in antibody diluting buffer g was added to each well, and the plates were incubated for 30 min before washing. This was followed by the addition of an optimized dilution of goat anti-mouse immunoglobulin G conjugated to horseradish peroxidase h in antibody diluting buffer to each well, and the plates were incubated for 30 min. After incubation and washing, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution i was added. After a 15-min incubation, the reaction was stopped with the addition of stop solution. j The OD at A450 was determined using an ELISA reader. k The inhibition of mAb 17B7 binding after incubation of the antigen with each test serum was calculated as a percent inhibition of the mAb with reference negative serum, using the following formula: % inhibition (%I) = 100 − [(test serum OD × 100) ÷ (mean negative control OD)]. The test serum was considered to be antibody-positive if the %I was ≥35%. The OIE-prescribed SN assay, which is a complement-enhanced microtiter SN assay for measuring EAV-neutralizing antibodies in equine serum, was performed as described previously. 19
Modification and improvement of cELISA
The mAb was changed from 4B2 to 17B7. The method of EAV antigen production was changed from propagation in rabbit kidney (RK)-13 cells 6 to primary equine spleen (ESP) cells as described above. The storage method of ELISA plates coated with EAV antigen was changed to a dry storage method for longer stability and to enable larger lot production. Serum incubation time was optimized. Other format variables including antigen coating buffer, blocker, sample volume, serum incubation time, and washing conditions were also optimized as described in the cELISA methods section above.
Statistical analysis of the correlation between cELISA and SN results
Correlation coefficient (r) and statistical significance (p-value) were determined to measure the strength and the direction of a relationship between results from a SN titer (log2) and cELISA result (%I) using Spearman rank correlation analysis. All statistical analyses were performed using R software (http://www.r-project.org/).
Results
Improved specificity and sensitivity of mAb 17B7-based cELISA and correlation with SN results
The mAb 4B2-based cELISA previously reported 6 was optimized by changing variables in the cELISA procedure including the cell line used to produce antigen, serum incubation time, the antigen coating procedure, the wash buffer, and monoclonal antibody for epitope competition with EAV-specific equine serum antibodies. The time of serum incubation for this mAb 17B7-based cELISA was adjusted to 2 hr based on examination of various incubation times using well-characterized SN-positive and SN-negative serum panels (Fig. 1).
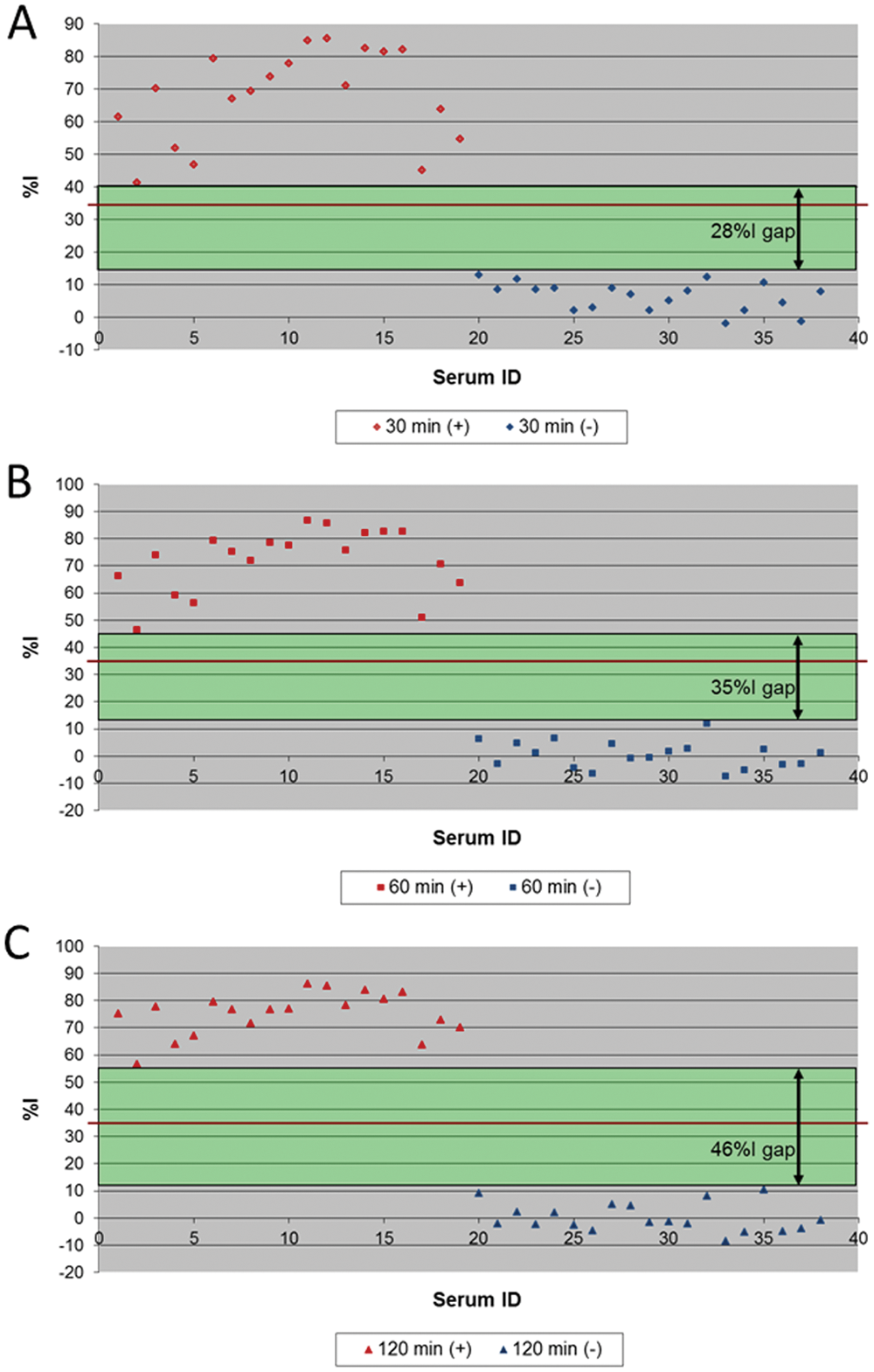
Longer serum incubation time in monoclonal antibody 17B7-based competitive enzyme-linked immunosorbent assay increased the resolution between serum neutralization (SN)-positive and SN-negative sera:
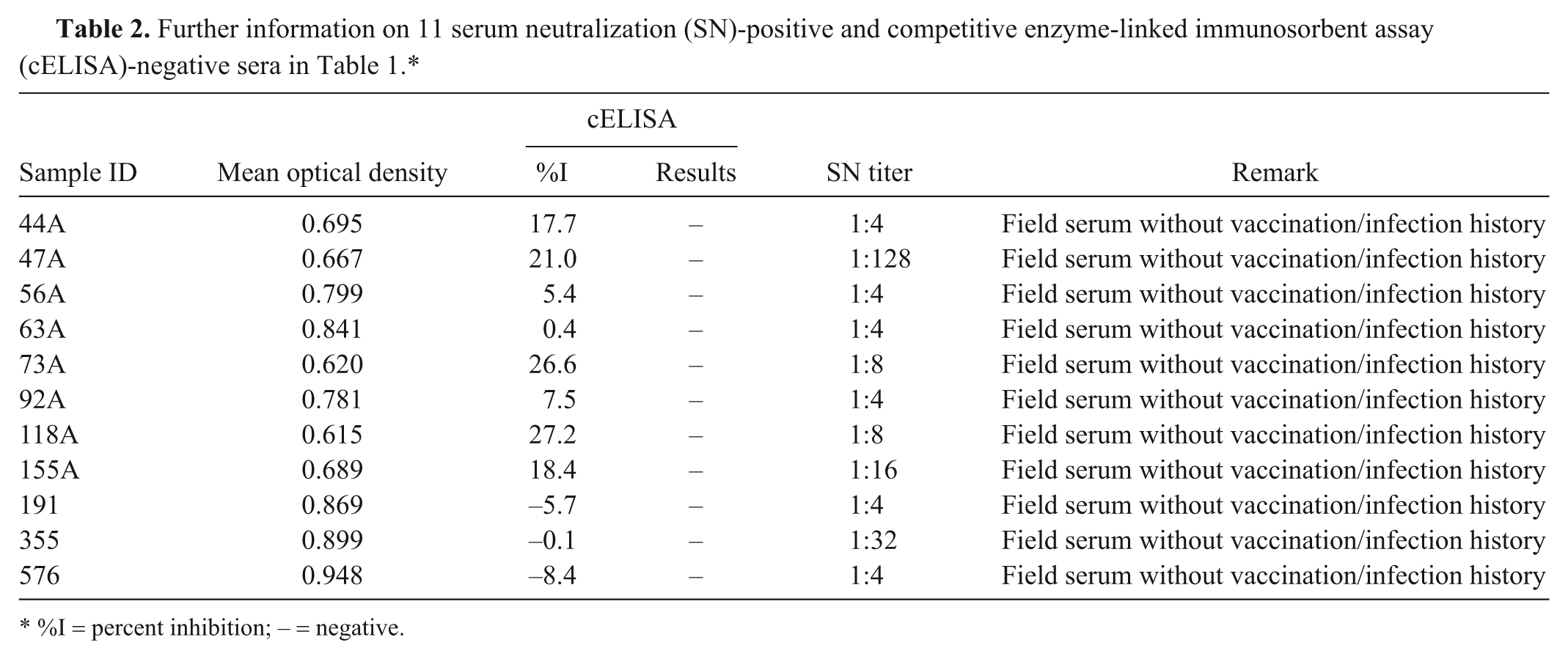
Such optimization resulted in an improved performance of the cELISA because there was a wider zone between the %I obtained with positive and negative sera (Fig. 2), which eliminated the need for an inconclusive range used in the previous mAb 4B2-based cELISA. 6 When the optimized mAb 17B7-based cELISA was compared with the SN assay results for 2,469 sera, the relative sensitivity was 95.5% and the specificity was 99.8% when ≥35%I (cutoff) was considered positive (Table 1). Of 11 sera with SN-positive but cELISA-negative results, 9 sera excluding 2 sera with 1:128 and 1:32 in SN titer had lower SN titers than 1:16 (Table 2). Further, there were no prozone reactions detected, and all of the 189 high SN-positive sera (≥1:256) were positive in the mAb 17B7-based cELISA without dilution of serum (Fig. 3). The results from the mAb 17B7-based cELISA demonstrated moderate, but highly significant (P < 0.0001) positive correlations with the results of the SN assay (correlation coefficient 0.51).
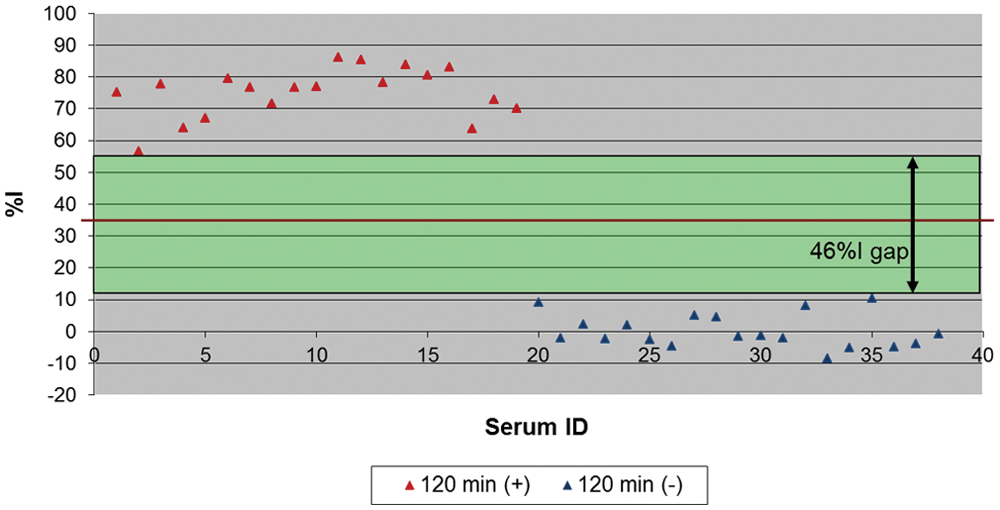
The resolution of monoclonal antibody 17B7-based competitive enzyme-linked immunosorbent assay. Red line shows the cutoff. %I = percent inhibition.
Diagnostic performance of the improved monoclonal antibody 17B7-based competitive enzyme-linked immunosorbent assay (cELISA).*

Sensitivity 95.5% and specificity 99.8% based on the comparison with serum neutralization result. + = positive; − = negative.
%I = percent inhibition; − = negative.
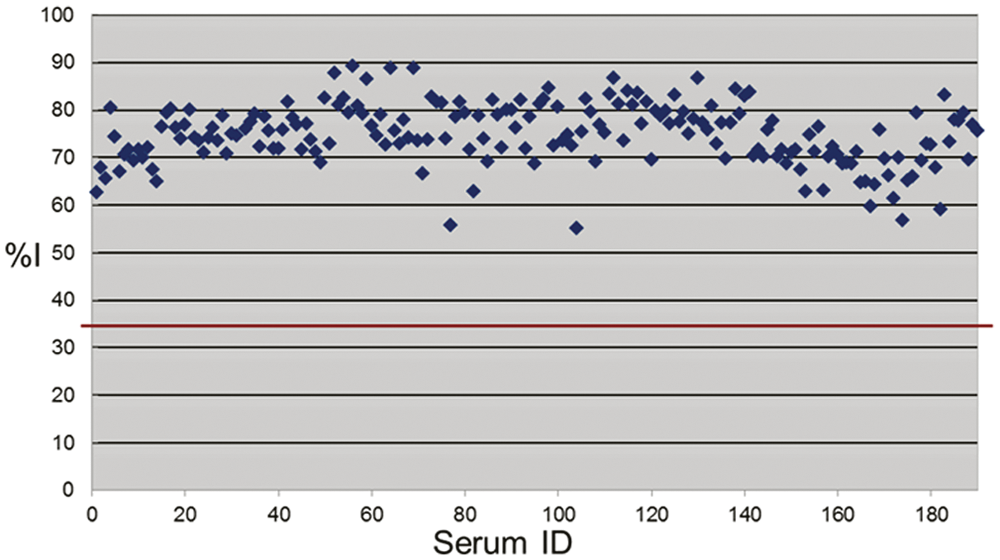
Percent inhibition (%I) of 189 sera with serum neutralization titer ≥1:256. Red line shows the cutoff.
Performance in cELISA with horse sera having non–EAV-specific cytotoxicity in SN assay
To evaluate the performance of the newly developed mAb 17B7-based cELISA, 70 horse sera having nonspecific cytotoxicity in the SN assay, 31 EAV seronegative and 39 EAV seropositive, were analyzed by cELISA. When the cELISA results were compared with SN assay results (Table 3), there was a 100% concordance between the 2 assays.
Concordance between the improved monoclonal antibody 17B7-based competitive enzyme-linked immunosorbent assay (cELISA) and the serum neutralization (SN) assay results using horse sera with cytotoxicity in SN assay.*
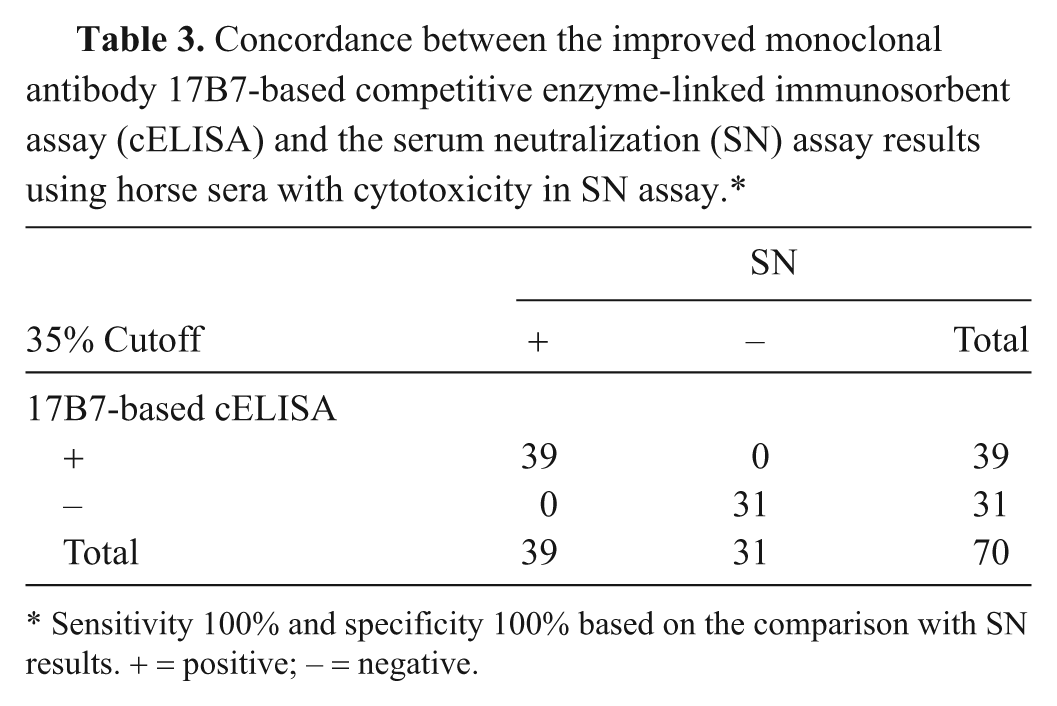
Sensitivity 100% and specificity 100% based on the comparison with SN results. + = positive; − = negative.
Discussion
In the current study, the cELISA was improved by modifying the competing gp5-specific mAb and other variables of the cELISA format from a previously developed assay. This improved cELISA reliably detected EAV-specific antibody responses from EAV vaccinated and infected horses. The sensitivity of the new mAb 17B7-based assay was 95.5% and the specificity was 99.8%. This sensitivity and specificity was obtained without prozones, which caused some high SN-positive sera to be cELISA negative without dilution, and without the need for a suspect range used for the mAb 4B2-based cELISA in a previous report. 6 In the initial study of the cELISA using mAb 4B2, the suspect range was 30–50%I, and sera in the suspect range included 7.9% of the SN-negative sera and 21.4% of the SN-positive sera (12% of total samples). 6 The lack of need for a suspect range and the absence of prozones using the new 17B7-based EAV cELISA are significant improvements.
In the initial development of the EAV cELISA, multiple mAbs including 17D3 specific to N and 4B2 specific to gp5 were examined, and the cELISA using mAb 4B2 was found to be more sensitive than that using mAb 17D3. 6 Initially, non-neutralizing mAb 17B7 specific for EAV gp5 was not pursued in the cELISA format. Monoclonal antibody 17B7, although non-neutralizing, is reported to have a reciprocal competition for epitope binding with mAb 4B2, suggesting close proximity of binding epitopes. 9 The major change in the improved cELISA was replacing the mAb from neutralizing mAb 4B2 to non-neutralizing mAb 17B7 to compete with EAV-specific horse serum antibody binding to its epitope on EAV gp5. Another format change was the propagation of EAV for ELISA antigen in ESP cells instead of RK13 cells. This change may have eliminated the binding of non-EAV antibodies to RK13 cell debris in antigen-coated wells, which could have sterically inhibited binding of the mAb. Subsequently, other variables in the cELISA format including serum incubation time, and buffers were changed to optimize the new mAb 17B7-based cELISA. The relative specificity of the new mAb 17B7-based cELISA was 99.8% when evaluated with 2,223 SN-negative sera, and the relative sensitivity was 95.5% with 246 SN-positive sera. To the authors’ knowledge, these results represent the best performance data to date among published EAV antibody-detecting ELISAs.4,6,10,13-15,18 The calculations of the sensitivity and specificity were based on the assumption that all SN results are correct. However, incorrect SN results due to nonspecific cytotoxicity and interlaboratory or inter-run variations have been reported6,19 and were in fact encountered in the field study using the mAb 17B7-based cELISA (unpublished data). When cELISA and SN results from 70 horse sera with non–EAV-specific cytotoxicity in the SN assay were analyzed, there was a 100% concordance between the cELISA and SN results, which further supports the advantage of this new cELISA. The mAb 17B7-based cELISA also eliminated the prozone reactions noted with the mAb 4B2-based cELISA 6 as none of 189 sera that had an SN titer ≥1:256 were negative when tested undiluted in the improved cELISA. Further, the improved mAb 17B7-based cELISA had a gap of 46%I between the lowest %I with SN-positive sera and the highest %I with SN-negative sera when tested with panels of well-characterized positive and negative sera (Fig. 2). This was in contrast to the results using the mAb 4B2-based cELISA on the same serum panel where there were overlaps between the lowest %I with SN-positive sera and the highest %I with SN-negative sera resulting in a lower resolution of the assay (data not shown). Therefore, the mAb 17B7-based cELISA demonstrated a significant improvement in resolution by eliminating overlaps in cELISA %I of SN-positive and SN-negative sera.
The moderate, but highly significant (P < 0.0001) correlation between SN results and cELISA results using the neutralizing mAb 4B2-based cELISA was somewhat expected. However, the moderate, but significant positive correlation between SN results and the results using the non-neutralizing mAb 17B7-based cELISA was surprising. That cELISAs using both mAbs correlate with an SN titer could be due to the similar topography of the 2 epitopes on gp5 even though the 2 mAbs differ with respect to neutralizing capacity. 9 The inability of mAb 17B7 to neutralize EAV may also correlate with lower immunological pressure for antigenic variation in this region, which might be beneficial in terms of diagnostic sensitivity. The EAV gp5 contains multiple neutralizing epitopes inducing immunodominant responses in EAV-infected horses.1,9 The sequences of immunodominant EAV gp5 are highly variable among isolates21,22 suggesting that the gp5 epitopes with immune pressure from neutralizing antibodies might vary more quickly and create problems for single epitope-based cELISAs. However, basing the cELISA on a conserved non-neutralizing epitope like the one recognized by mAb 17B7 may alleviate these problems and allow detection of both EAV-infected and vaccinated horses.
In conclusion, the improved mAb 17B7-based cELISA had a specificity and sensitivity of 99.8% and 95.5%, respectively, when compared with SN assay results. The modified cELISA improved the resolution of SN-negative and SN-positive samples near the cutoff eliminating a suspect range, had no prozone reactions, and there were no problems caused by sera with non-EAV cytotoxicity in the SN assay. Pending further field validation, this improved cELISA may become a suitable alternative to the SN assay.
Footnotes
Acknowledgements
The authors thank Amanda Grimm, Chandima Bandaranayaka-Mudiyanselage, Lorraine Tanaka, Sara Schlee, and LaDawn Baker for technical support.
a.
ARVAC, Fort Dodge Animal Health, Fort Dodge, IA.
b.
ARVAC, Fort Dodge Animal Health, Fort Dodge, IA.
c.
Sigma-Aldrich, St. Louis, MO.
d.
High binding 96-well plates, Costar Stripwell (Costar 2592), Vernon Hills, IL.
e.
ELISA blocking buffer, VMRD Inc., Pullman, WA.
f.
ELISA antibody diluting buffer, VMRD Inc., Pullman, WA.
g.
IMPAK Co., Los Angeles, CA.
h.
Goat anti-mouse immunoglobulin G conjugated to horseradish peroxidase, VMRD Inc., Pullman, WA
i.
TMB substrate solution, SurModics Inc., Eden Prairie, MN.
j.
SurModics Inc., Eden Prairie, MN.
k.
Multiskan MCC/340 ELISA reader, Titertek Instruments Inc., Huntsville, AL.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the general research funding of VMRD (Veterinary Medical Research and Development) Inc.