
Abstract
A real-time reverse transcription polymerase chain reaction assay (PCR test) based on genome segment 10 of Bluetongue virus (BTV) was developed. The PCR test consists of robotized viral RNA isolation from blood samples and an all-in-one method including initial denaturation of genomic double-stranded RNA, reverse transcription polymerase chain reaction (RT-PCR), and real-time detection and analysis. Reference strains of the 24 recognized BTV serotypes, isolates from different years, and geographic origins were detected. Other orbiviruses such as African horse sickness virus, Epizootic hemorrhagic disease virus, and Equine encephalosis virus were not detected. Experimentally infected animals were PCR positive from 2 days postinoculation, which was earlier than fever, other clinical signs, or seroconversion. The diagnostic sensitivity and specificity were very close to or even 100%. The PCR test played a key role in the detection of BTV serotype 8 in August 2006 in The Netherlands. The outbreak in a completely naive ruminant population allowed for further evaluation of the PCR test with field samples. In 2006, the correlation between enzyme-linked immunosorbent assay and PCR results was estimated to be 95%. In the following years, the PCR test was used for diagnosis of diseased animals, for testing of healthy animals for trade purposes, and for detection of BTV RNA in different species of the insect vector, Culicoides. In the autumn of 2008, BTV serotype 6 unexpectedly emerged in northwest Europe and was also detected with the PCR test developed in the current study. The performance in routine use over 5 years has been recorded and evaluated.
Introduction
Bluetongue virus (BTV; family Reoviridae, subfamily Sedoreovirinae, genus Orbivirus) infects a wide range of ruminants and is responsible for significant losses of sheep and goats. Bluetongue is an arthropod-borne disease, and transmission of BTV occurs by bites of “vector-competent” species of Culicoides. 26 Bluetongue has a worldwide distribution between the latitudes of 35°S and 40°N but can expand significantly, up to 50°N depending on local conditions. 20 Since 1998, BTV has invaded European countries around the Mediterranean basin 27 and was therefore classified as an emerging threat for livestock at higher latitudes (Elbers A, van Rijn PA, van Rooij EMA: 2003, Report: Aanzet tot een risk analysis m.b.t. introductie van bluetongue virus en West Nile virus in Nederland [Towards a risk analysis regarding the introduction of bluetongue virus and West Nile virus in the Netherlands]. Available at: http://www.cvi.wur.nl/NR/rdonlyres/CC4EC0FD-3F41-4C98-A2A5-E08903091408/11385/RiskAssessmentBT.pdf. In Dutch). Bluetongue virus serotype 8 invaded several northwestern European countries starting in 200612,18 and, in addition to sheep and goat, also caused serious illness in cattle. 4 The epidemiology of bluetongue in Europe has been affected by the presence of vectors different from Culicoides imicola, which are possible competent vectors in areas with a moderate climate.11,24,25 The huge outbreak and expansion of areas affected by other BTV serotypes highlight the need for rapid, easy, and reliable diagnostics for all BTV serotypes.
Bluetongue virus includes 24 recognized serotypes, 9 but several new serotypes are proposed.16,21 The BTV genome consists of 10 genome segments of double-stranded RNA (dsRNA; S1–10), which display remarkable genetic variability. Many molecular tests based on reverse transcription (RT) and polymerase chain reaction (PCR) have been developed that target different genome segments of BTV. 15 The tests meet many requirements for molecular diagnostics, but some have disadvantages with regard to development for high-throughput routine diagnostics. 5 Performance in practical use over long-term periods has not been described.
In the current study, the development, validation, and practical use of a PCR test over a period of 5 years is described. The PCR test targets 2 highly conserved regions of genome segment S10 and combines robotized isolation and an all-in-one method including initial denaturation of dsRNA, RT, PCR, and real-time detection and analysis.
Materials and methods
Viruses
Except for BTV8/net06 and BTV6/net08, all BTVs and related orbiviruses of the following serogroups—African horse sickness virus (AHSV), Epizootic hemorrhagic disease virus (EHDV), and Equine encephalosis virus (EEV)—were purchased and then passed once in BHK (baby hamster kidney)–21 cells according to standard procedures. BTV8/net06 and BTV6/net08 were isolated in The Netherlands in 2006 and 2008, respectively (http://www.reoviridae.org/dsRNA_virus_proteins/), on embryonated chicken eggs (ECE) 14 and subsequently passed according to standard procedures. Virus stocks were stored at −70°C. Virus dilutions in medium were used in the PCR test and for experimental infections of animals. Occasionally, ethylenediamine tetra-acetic acid–treated whole blood (EDTA-blood) of infected cattle or sheep was used for inoculation of naive animals after washing without freezing or after storage in aliquots at −70°C.
Virus isolation
Virus isolation (VI) was carried out on ECE or by passages on monolayers of BHK-21, Vero (African green monkey kidney epithelial), or KC (Kenyon Culicoides) cells according to standard procedures. 30 After 1 passage on ECE, homogenates were prepared and tested in the PCR test or passed in BHK-21, Vero, or KC cells.
Animal experiments
Animal experiments were performed in cattle, sheep, and goats. Animals were monitored daily for clinical signs and sampled frequently for EDTA-blood and clotted blood (serum). Samples were divided into suitable aliquots and stored at 4°C for ≤2 days or at −70°C (EDTA-blood) and at −20°C (serum) for longer periods. All experiments with live animals were performed under the guidelines of the European Community (86/609) and were approved by the Committee on the Ethics of Animal Experiments of the Central Veterinary Institute.
Experiment 1
Two groups of 6 sheep were used. All sheep were negative for BTV RNA and BTV antibodies as determined by the PCR test and enzyme-linked immunosorbent assay (ELISA), respectively. Five sheep per group were intravenously inoculated with 1 ml of 105 TCID50/ml of BHK-21–adapted BTV serotype 2 or 4 of the panel of reference BTV strains of the Institute for Animal Health, Pirbright, United Kingdom (IAH-Pirbright, U.K.) (http://www.reoviridae.org/dsRNA_virus_proteins/). Animals were sampled frequently for EDTA-blood and serum. After 70 days postinoculation (dpi), the experiment was terminated.
Experiment 2
Three of 5 sheep and 2 of 4 goats, all negative for BTV and BTV antibodies, were inoculated intravenously with BTV8/net06. After 44 days, the experiment was ended by euthanizing the remaining animals. Detailed descriptions of clinical signs have been described. 3
Experiment 3
In 2006, 3 naturally infected pregnant heifers were used as donors of BTV serotype 8. These heifers had no clinical signs but were positive for serotype 8 and serotype 8 antibodies, and they were estimated to be infected at least 50 days before the first sampling. One hundred milliliters of EDTA-blood was collected from each heifer. Total pooled cell fractions were washed several times with phosphate buffered saline (PBS), resuspended, and pooled in a total volume of 300 ml (washed EDTA-blood). Three recipient cows negative for BTV and BTV antibodies were inoculated with 70 ml intravenously and 30 ml subcutaneously. After 7 and 21 dpi, EDTA-blood was harvested from 2 of 3 inoculated cows identified as BTV positive by PCR and was stored at −70°C (EDTA-blood). Recipient cows and heifers were monitored and sampled weekly until the end of the experiment (approximately 150 and 200 dpi, respectively).
Experiment 4
EDTA-blood collected in experiment 3 was used for inoculation. Seven of 15 pregnant cows, PCR and ELISA negative for BTV, were inoculated with 20 ml intravenously and 20 ml subcutaneously. Detailed descriptions of clinical signs have been described. 4
Field samples
For the current study, coupled samples of EDTA-blood and serum (taken from the same animal on the same day) were collected in the autumn of 2006 and early 2007. Coupled samples were stored in suitable aliquots at 4°C for ≤2 days before testing and for longer periods at −70°C for EDTA-blood and at −20°C for serum.
Serology
The presence of BTV antibodies in sera of experiment 1 was investigated by the ELISA developed at IAH-Pirbright, U.K. 1 Sera from experiments 2–4 and from the field were tested by ELISA according to the instructions of the supplier. a
PCR test
Viral dsRNA was isolated by a robot with a capacity of 32 samples per isolation run. b In a later stage, a robot with a capacity of 96 samples per run was implemented. b From 100 µl of EDTA-blood diluted with PBS (1:1), RNA was isolated according to standard procedures using a commercial kit. c RNA was eluted in 50 µl RNase-free H2O. A representative isolation run of 32 samples contained 6 controls, resulting in a final capacity of 26 test samples per isolation run. Three negative controls (which consisted of 200 µl PBS) and 3 positive controls were part of each isolation run. Positive controls contained different dilutions of BTV grown on BHK-21 cells. The weak positive control (cutoff value) contained the highest dilution of virus with a reproducible positive signal. The strong positive control was the highest dilution of virus resulting in a maximal ratio of optical density (OD)530/OD640. The third positive control (intermediate control) was a dilution of virus with a result between the weak and strong positive control (Fig. 1). Samples of 1 isolation run, including positive and negative controls, were immediately tested or stored at −70°C. Field samples were always tested in the real time RT-PCR assay together with the controls of the same isolation run.
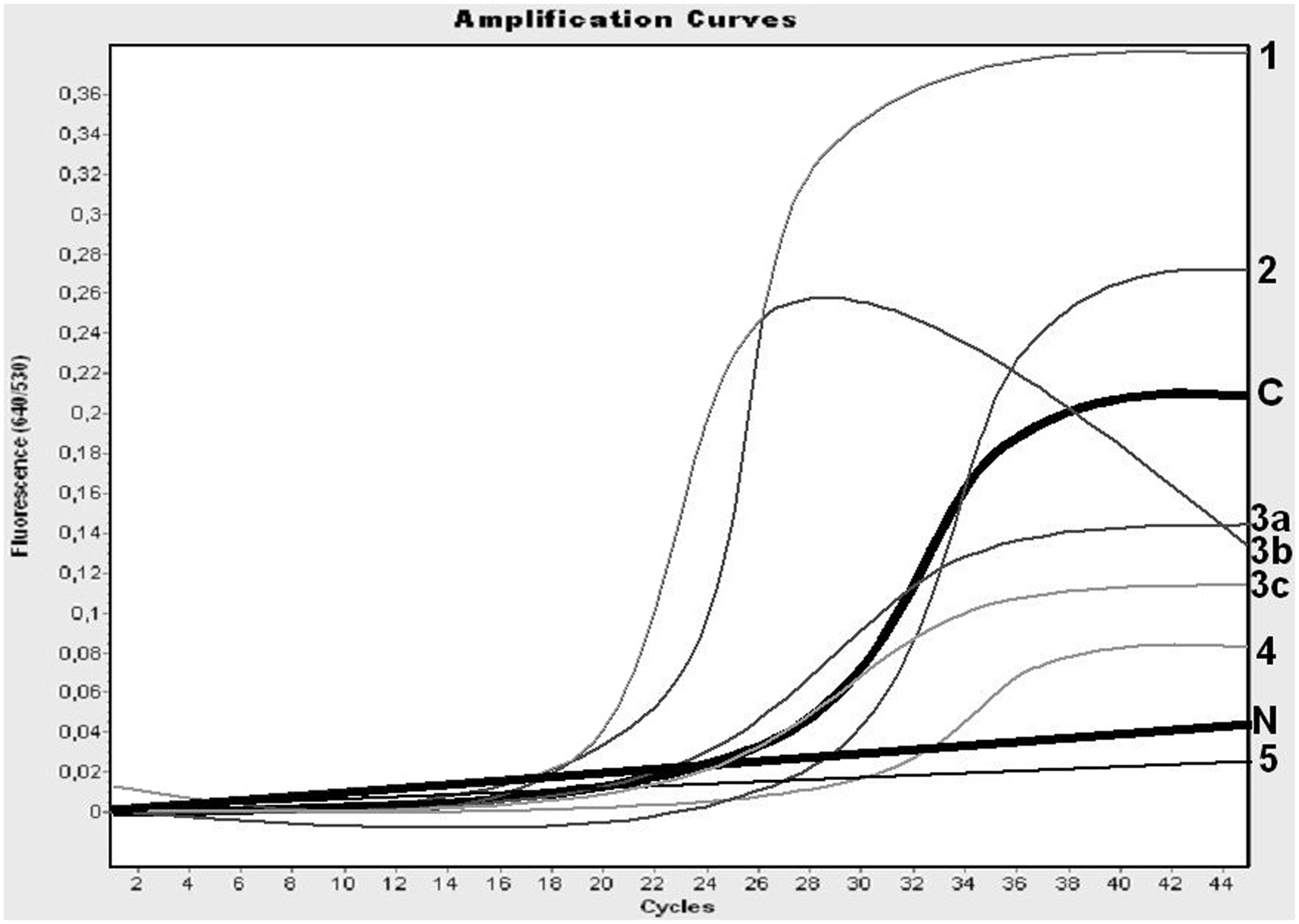
Interpretation of polymerase chain reaction (PCR) results of field samples. The negative control (N) and the curve of the cutoff (C) are presented by thick lines. The PCR results interpreted as positive (1, 2, 3a, and 3b), as inconclusive (3c and 4), and as negative (5) are presented. Note: the amount of RNA in the sample of the weak positive control was chosen relatively high to avoid false-positive interpretations, and the interpretation of “negative” is limited for results very similar to the negative control to avoid false-negative interpretations.
Primers and probes were designed by alignment of available sequences, including sequences in GenBank and provided by others prior to publication. Primers and probes were designed by selection of conserved regions in genome segments S5, S8, and S10. Initial denaturation of dsRNA, first-strand complementary DNA (cDNA) synthesis (RT), PCR, and real-time monitoring were performed in a closed capillary. Real-time RT-PCR was performed using a thermal cycler. b In a later stage, real-time RT-PCR was also performed in a different thermal cycler with a 96-sample format. b Forward primer 5′-AGTGTCGCTGCCATGCTATC-3′ and reverse primer 5′-GCGTACGATGCGAATGCA-3′ were used. Amplicons were detected by probe 5′-6FAM-CGAACCTTTGGATCAGCCCGGA-XTamra. One reaction consisted of 0.25 µM of the forward and reverse primer, 0.25 µM of the probe, 2.75 mM of MnCl2, 7.5 µl of a commercial master mix, d and 5 µl of RNA in a total volume of 20 µl. Thermocycling conditions were as follows: 20 sec at 98°C, 20 min at 61°C, 30 sec at 95°C, 40 cycles (1 sec at 95°C, 10 sec at 61°C, 15 sec at 72°C) followed by 30 sec at 40°C, and storage at 4°C. Amplification was monitored in real time by the ratio of the OD530 and OD640 (OD530/OD640) using commercial software. b A PCR test was considered valid if all negative controls were negative and positive controls were “positive” (Fig. 1). In case of an invalid PCR test, the entire set of isolated RNA was tested again.
Field samples with crossing point (Cp) values calculated by the software b and with an S-shaped curve of OD530/OD640 values at least partly higher than those of the cutoff value were interpreted as “positive” (Fig. 1). Results were interpreted as negative in cases of no significant increase in the OD530/OD640 value. All other results were interpreted as inconclusive (Fig. 1). Samples with inconclusive results were tested again by repeating the real-time RT-PCR assay together with the isolated RNA of the positive and negative controls from the same isolation run. Alternatively, RNA was isolated from a new aliquot of the respective EDTA-blood sample in a new isolation run, including positive and negative controls, to repeat the entire procedure of the PCR test.
Results
Development of the PCR test
Prototypes of RT-PCR assays were developed that theoretically detected all BTV strains of which the sequences of the PCR targets were available (in silico sensitivity). Sequences of other relevant serogroups of orbiviruses (EHDV, AHSV, and EEV) were used to confirm the selectivity of primers and probes (in silico specificity). Ten prototype assays were studied with a representative panel of 24 serotypes of BTV as provided by IAH-Pirbright, U.K. Four of the prototypes were further studied with EDTA-blood samples from experiment 1. The most promising prototype was based on genome segment S10, and this assay was selected for extensive optimizing and validation. The optimal conditions were defined as the earliest Cp values combined with the maximal OD530/OD640 ratio. The robustness of the PCR test was studied by deliberate deviations of volumes, temperatures, and time intervals. The volumes of premix (15 µl) and template RNA (5 µl) may vary up to 2 µl without reduction of sensitivity. Annealing temperature at 61 ± 2°C and variation in time intervals (±1 sec) did not affect the sensitivity. Because the template for RT is mainly viral dsRNA, special attention was taken on the denaturation of dsRNA before synthesis of the first strand of cDNA in the presence of all reagents. Maximal sensitivity was achieved by initial denaturation at 98°C for 20 ± 5 sec. Furthermore, for practical reasons, it was confirmed that isolated RNA could be retested without loss of sensitivity after storage up to 7 days at 4°C, or after up to 10 freeze–thaw cycles. New batches of isolation and amplification kits, primers, probe, and different isolation robots and different thermal cycler machines were implemented in a 5-year period of study. The relative standard deviation of the repeatability of the strong and weak positive control was 0.50% and 2.03%, respectively (Fig. 2). The relative standard deviation of the reproducibility of the strong and weak positive control was 1.9% and 2.3%, respectively.
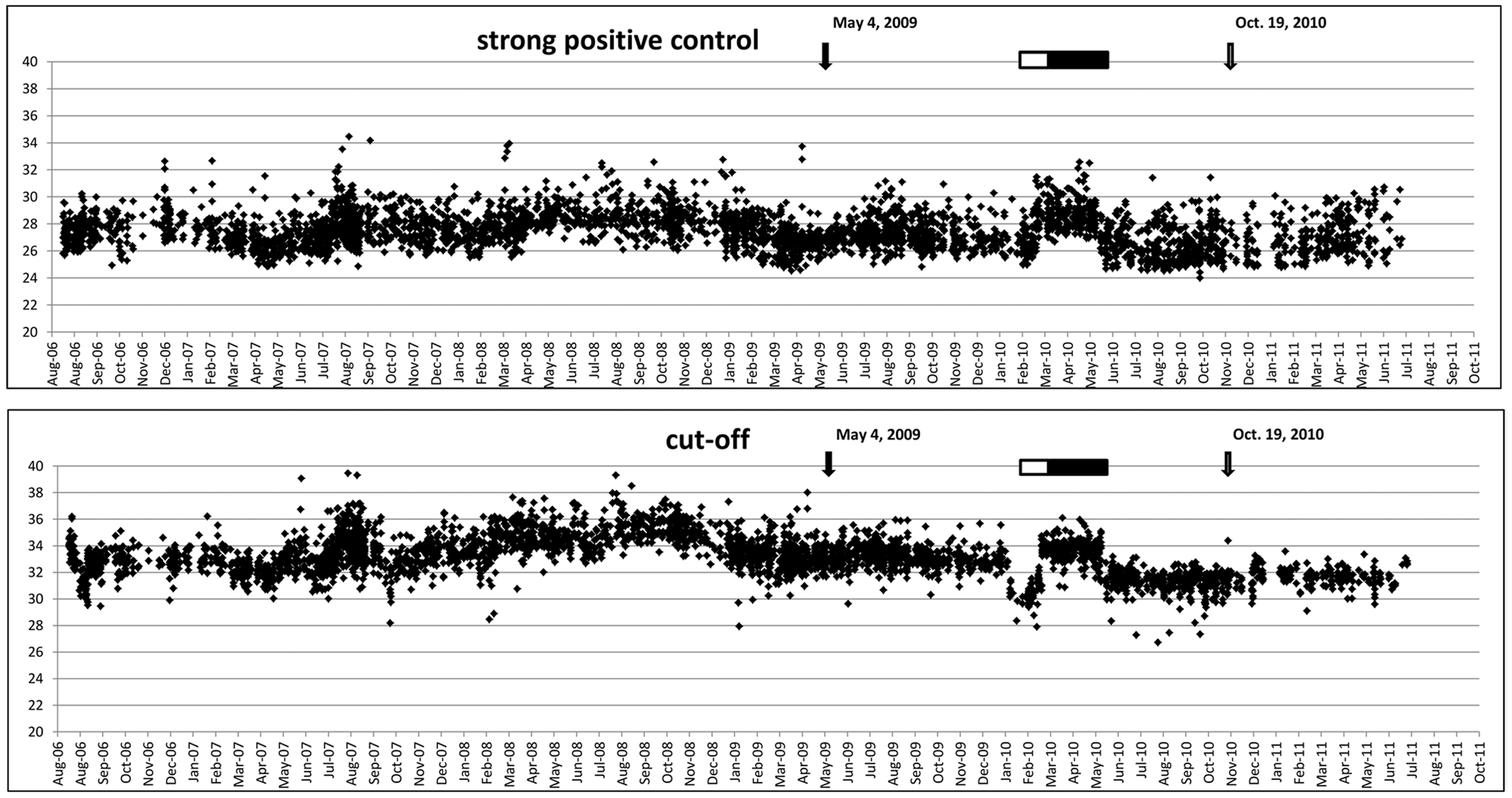
Records of the crossing point values of the strong positive control and the cutoff over the 5-year period of practical use. Implementation of the isolation kit, amplification kit, primers, probe, equipment, and personnel was also recorded. Scaling to the 96-sample polymerase chain reaction (PCR) system (top) and 96-sample format isolation robot (bottom) are indicated. Significant differences in performance associated with the implementation of new batches of kits or primer/probes are indicated by an open and filled bar, respectively.
Sensitivity
Desktop selection of conserved regions for primers and probe by alignment of known sequences of BTVs was the first step to develop a PCR test with a high theoretical or in silico sensitivity. Selected conserved regions for primers and probe in S10 of BTV have a maximum of 1 mismatch (data not shown).
Twenty-four BTV serotypes from cell cultures, including isolates from Australia, the United States, southern Europe, northwest Europe, and South Africa, were detected (in vitro sensitivity). The correlation between virus titer and PCR signal was found between 3.3 and 3.5 Cp/log dilution (R2 of >0.995) for the panel of reference BTV strains over a wide range of dilutions, indicating a comparable detection limit for all BTVs. Virus detection by PCR was more sensitive than VI on ECE (results not shown).
The detection of BTV by PCR in animals (diagnostic sensitivity or in vivo sensitivity) was investigated with samples from 4 animal trials in which different BTV strains were used. Two of these animal trials, experiments 2 and 4, have been published.3,4 No severe clinical signs were observed in experiments 1 and 3, except fever (>40°C) at 1 or more days in the first 2 weeks postinfection in experiment 1. In both groups, 1 of 5 inoculated animals became PCR inconclusive late after infection, at 21 or 32 dpi, respectively, followed by successive PCR-negative results. All other animals remained PCR positive up to 70 dpi, the end of the experiment. In experiment 3, 3 naturally infected heifers remained PCR positive to >140 dpi and became PCR negative after 2 consecutive weekly samples with PCR-inconclusive results. Two experimentally infected heifers became PCR positive at 5 and 7 dpi and became PCR negative at approximately 170 dpi after 2 consecutive weekly samples with PCR-inconclusive results. The third recipient remained PCR and ELISA negative, indicating an unsuccessful experimental infection.
In summary, 29 animals infected with different BTV serotypes were all detected by the PCR test at an average of 4 dpi. Infection was subsequently confirmed by ELISA for BTV antibodies and by repeated testing by PCR. In 1 case, experimental infection was unsuccessful as determined by successive negative PCR tests. This animal also remained ELISA negative, confirming the unsuccessful infection. In conclusion, the range of detection for all BTVs was comparable, and the diagnostic sensitivity in this study was approaching 100%.
Specificity
Alignment of primer and probe sequences to all known sequences, including those of genetically related viruses, was performed to determine the theoretical selectivity or in silico specificity. Each of the primers and probe did not match to EHDV, AHSV, or EEV sequences or to any other known sequences submitted to GenBank, meaning ≥4 mismatches per primer or probe were found (not shown).
The analytical or in vitro specificity of the primers was studied in more detail by 2 methods, dye detection e and/or agarose gel electrophoresis. No amplification was observed for AHSV, EHDV, and EEV showing a high selectivity for amplification (not shown). Selectivity was further achieved by real-time detection/analysis with the developed selective probe for BTV amplicons.
The diagnostic or in vivo specificity was determined by testing EDTA-blood samples from BTV-free animals. Eighteen cattle, 17 sheep, and 4 goats served as negative control animals in trials and/or were sampled prior to infection. In total, more than 200 samples from 39 animals were ELISA and PCR negative (not shown). In experiment 1, 1 negative control sheep became PCR positive at day 21 and following samplings in the experiment. Indeed, this sheep seroconverted in ELISA 9 days later for the first time, confirming BTV infection much later via an unknown route. In conclusion, the diagnostic specificity of the PCR test was very close to or even 100%.
Practical use
The PCR test was used for field samples after the incursion of BTV serotype 8 in northwest Europe in August 2006. Bluetongue had never previously been reported in The Netherlands, and a nonvaccination policy against bluetongue was in place. Consequently, BTV infection could be detected by either PCR or ELISA, although antibodies could be detected later in the infection. Coupled samples of EDTA-blood and serum, taken on the same day from the same animal, collected during August–December 2006, were studied by ELISA and PCR tests (Table 1). For cattle, 369 of 403 coupled samples were both ELISA and PCR positive. All coupled samples (n = 230) of seropositive sheep were also PCR positive. None of the tested goats were PCR or ELISA positive in this period.
Diagnostic sensitivity in cattle and sheep.*
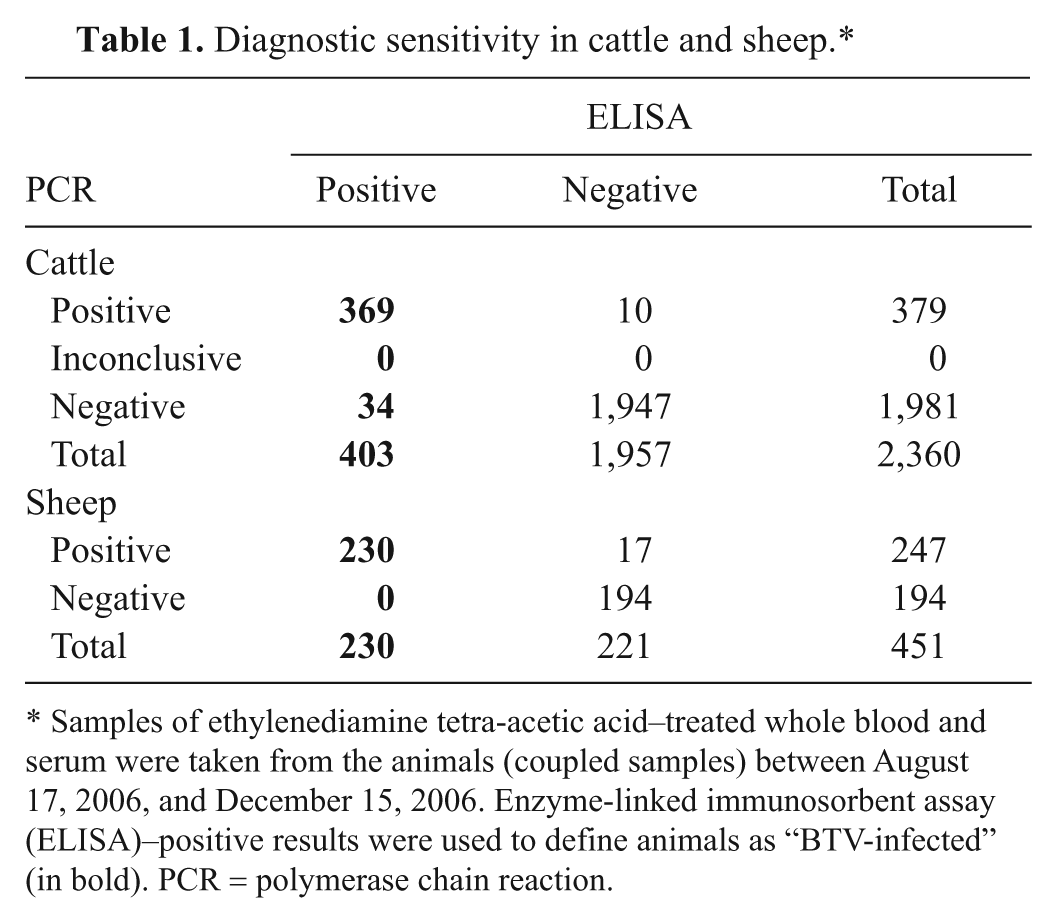
Samples of ethylenediamine tetra-acetic acid–treated whole blood and serum were taken from the animals (coupled samples) between August 17, 2006, and December 15, 2006. Enzyme-linked immunosorbent assay (ELISA)–positive results were used to define animals as “BTV-infected” (in bold). PCR = polymerase chain reaction.
Similarly, coupled samples were selected in the vector-free period between mid-December 2006 and the end of March 2007. Seronegative animals were expected to remain PCR negative in this period by the absence of BTV circulation. Indeed, all seronegative sheep (n = 543) and goats (n = 187) were also PCR negative (Table 2). For cattle, 1 of 247 seronegative animals was PCR positive. It appeared that this cow was sampled very soon after the official start of the vector-free period (December 27, 2006) and was likely an exceptional example of acute infection. In general, a very high correlation between ELISA and PCR results was found in 2006, the first year of BTV circulation (for positive results) and in the vector-free period (for negative results).
Diagnostic specificity in cattle, sheep, and goat.*
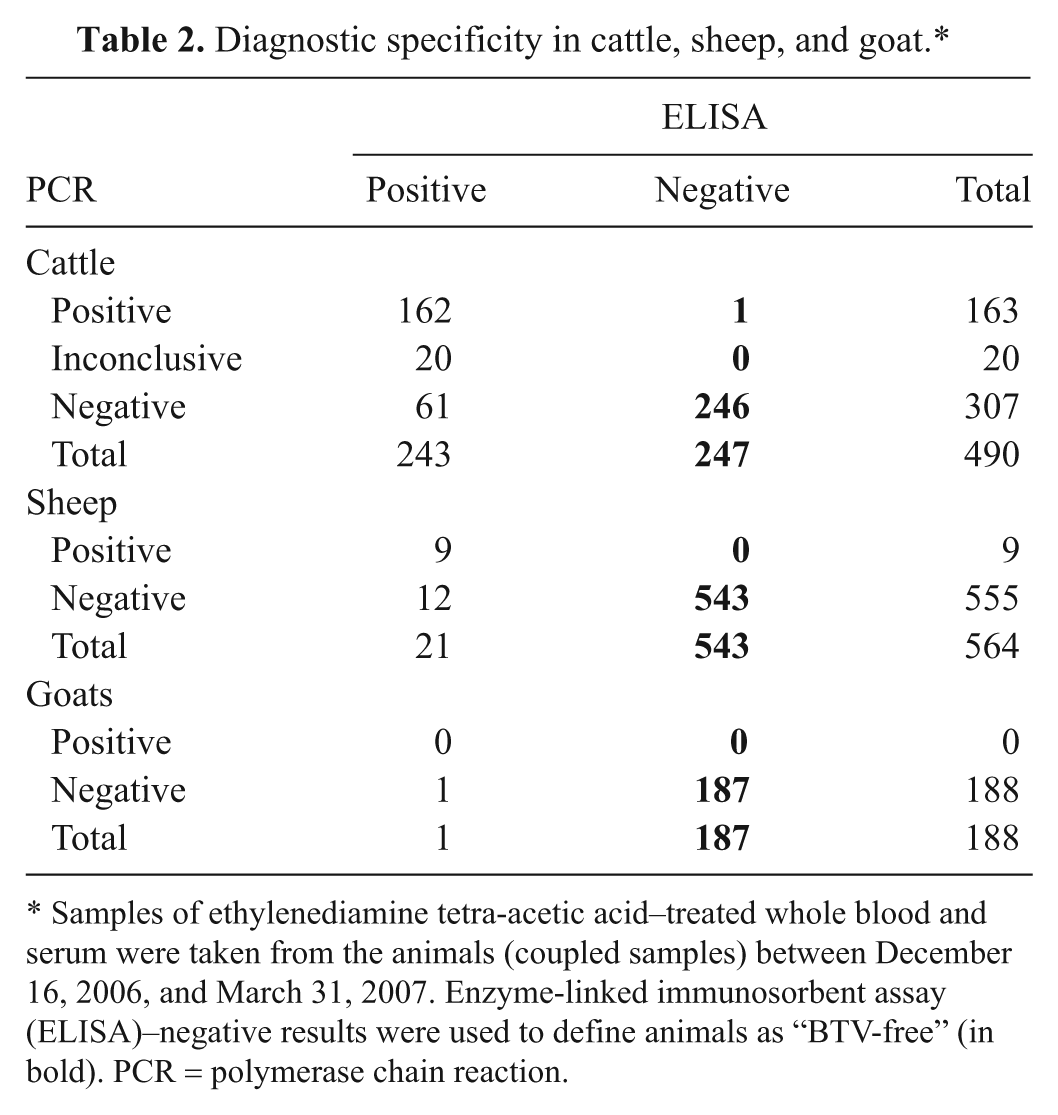
Samples of ethylenediamine tetra-acetic acid–treated whole blood and serum were taken from the animals (coupled samples) between December 16, 2006, and March 31, 2007. Enzyme-linked immunosorbent assay (ELISA)–negative results were used to define animals as “BTV-free” (in bold). PCR = polymerase chain reaction.
In 2007, seropositive animals could have been infected in the previous year. Therefore, comparative positive ELISA and PCR results were only possible in the first year of BTV circulation, starting with a completely naive ruminant population. ELISA positivity after natural infection lasts longer than PCR positivity and certainly overlaps periods of BTV circulation. Furthermore, starting in 2008, vaccination with an inactivated BTV serotype 8 vaccine was begun, and thus detection of infected animals by ELISA was no longer possible.
Regarding sustainability of the PCR test, primers and probe were annually checked by alignment to sequences in GenBank and other available sequence data sets (in silico sensitivity and specificity). Occasionally, new Orbivirus isolates were requested to check the in vitro sensitivity and specificity. Since 2006, test results of the annual ring trials organized by the Community Reference Laboratory for Bluetongue (IAH-Pirbright, U.K.) have been in full agreement with those of the organizing laboratory.
Over a 5-year period, the performance of the PCR test was continuously recorded during testing of samples to control the BTV serotype 8 epidemic. All negative controls were negative following the criteria for diagnosis (not shown). Sporadically, false-positive test results of negative controls were found, and subsequently the entire PCR test, RNA isolation, and RT-PCR assay were repeated as contamination was suspected. In general, positive controls showed limited variation. Occasionally, the PCR results of positive controls varied by >2 SD (“invalid”), and these runs were subsequently repeated. In the 5-year period of operation, change of reagents, equipment, and personnel was also recorded. No repeatable difference of >2 SD was observed in test performance; however, small variations (<2 SD) could be observed (Fig. 2). On 2 occasions, an obvious change in performance was associated with the implementation of a new batch of reagents (Fig. 2). A next step in high-throughput diagnostics was achieved by implementation of a 96-sample format system using the same details and conditions of the method described herein. In different periods, 96-sample format systems for isolation and for RT-PCR were implemented in the diagnostic capacity (Fig. 2). No difference and variability in test performance were noticed by this scaling up.
A summary of PCR results over 5 years of practical use in an epizootic situation followed by a monitoring period is shown in Figure 3. Inconclusive PCR results were observed to be in the majority after the peak of infections in 2006 and 2007. Positive PCR results for field samples were no longer found after 2008.
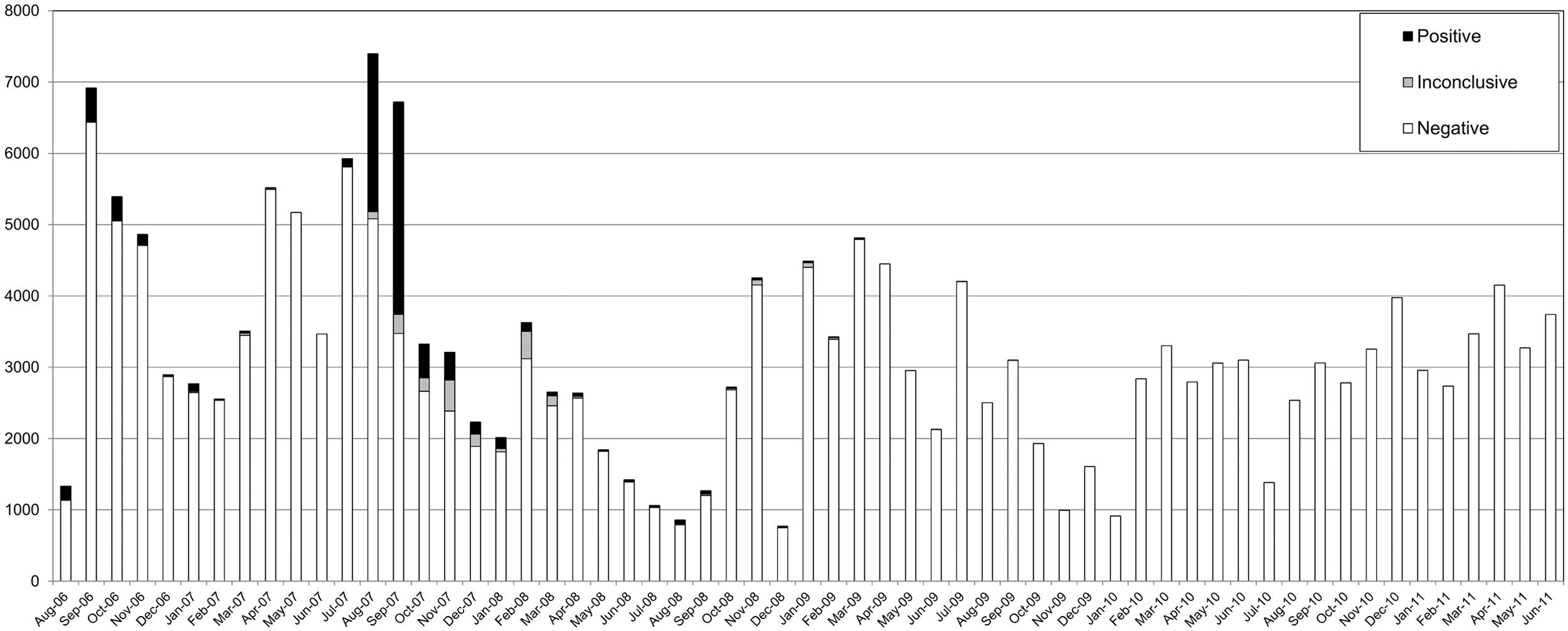
Overview of the monthly numbers of samples tested in the 5-year period. Reverse transcription polymerase chain reaction (RT-PCR) results are presented as positive, inconclusive, or negative in black, gray, or white bars, respectively. In total, approximately 200,000 samples were tested by PCR during the epizootic and post-epizootic period.
Discussion
As part of a preparedness program on bluetongue, a high-throughput PCR-based diagnostic system was developed to detect BTV. The PCR test provided early detection of all recognized BTV serotypes, as well as reliable and rapid testing of large numbers of samples.17,18 An average of over 3,000 samples per month (in total >200,000 samples) were tested in a 5-year period (Fig. 3). It has to be noted that Figure 3 does not reflect the number of BTV-infected animals but represents the number of tested EDTA-blood samples and is a strong underestimation of the actual number of BTV-infected animals. Furthermore, most samples from January 2009 onwards were tested for export purposes and originated from healthy cattle.
Polymerase chain reaction assays for BTV with manual RNA isolation have been described.2,6,28 Comparison between manual and automated RNA isolation has shown a top 3 position for automated RNA isolation for several previously developed PCR tests. 29 Manual RNA isolation is not considered practical to deal with epizootics in which large numbers of samples need to be tested. Previously published real-time PCR tests for BTV need initial denaturation of dsRNA before the addition of enzymes.8,15,19 The PCR test described in the current study includes initial denaturation of dsRNA, first-strand cDNA synthesis (RT), PCR, and detection/analysis by real-time technology in the presence of all reagents of the PCR test. This all-in-one method further reduces labor, as reopening of test tubes is no longer necessary. Other key benefits include avoiding reopening to minimize risk of accidents and cross-contamination, shortening the total operation time, and reducing costs. Implementation of a 96-sample format system for both dsRNA isolation and PCR for this all-in-one method has been accomplished (Fig. 3). The all-in-one method and the scaling to a 96-sample format system created a truly high-throughput system that will be very helpful in further automation of high-throughput diagnostics to control epizootics. 5
Sequences of virus isolates become available rapidly, even without virus cultivation.10,16 At present, the primers and probe contain at most 1 mismatch per oligo for all BTV strains. Furthermore, none of the mismatches are located near the 3′-end of PCR primers. In contrast, more than 4 mismatches are present in other orbiviruses, such as EHDV, AHSV, and EEV.
In terms of the onset of clinical signs, the ELISA and PCR test results in the current study are in agreement with other studies 13 and varied between animal trials. This variation is likely due to differences in species, status, and age of the animals, as well as by differences in dose, route of administration, origin, history, and the BTV strains used in the work.
The PCR results were interpreted as positive, inconclusive, or negative. An inconclusive area between the cutoff value of the PCR test and the negative control was defined to reduce the percentage of false positives and false negatives. On one hand, false-positive PCR results could incorrectly change the status of a BTV-free animal, farm, compartment, region, or country. As this could have serious consequences, the cutoff value of the PCR test was therefore chosen relatively high to lower the risk of an incorrect (positive) interpretation of PCR results. On the other hand, false-negative PCR results have undesirable consequences for bluetongue control, which include the national or international movement of infected animals to BTV-free areas. Depending on the impact, inconclusive PCR results were tested again and/or the respective animals were resampled. Animals must be tested PCR negative for national or international movement.
In 2006, due to the specific circumstances of this first year of BTV circulation, the status “recently infected by BTV” could be determined by ELISA and/or PCR tests. However, on some occasions, a fast BTV clearance after experimental infection was observed. Extrapolated to the field situation, a minority of ELISA-positive animals could have become PCR negative in August–December 2006 by fast BTV clearance (Table 1). For sheep, most of the animals tested were showing clinical symptoms and thus were acutely infected; therefore, all ELISA-positive sheep were PCR positive.
New infections (PCR+/ELISA–) were highly unlikely from December 2006 to March 2007 as this was the vector-free period in northwest Europe. Consequently, ELISA-negative animals in this period could be defined as “BTV-free.” Indeed, except for 1 animal, all ELISA-negative animals were also PCR negative (Table 2).
After the first period of BTV circulation, the percentage of PCR–/ELISA+ cattle became higher, and no inconclusive results were found in this period for sheep (Table 2). This is in agreement with the longer prolongation and slower decline of the PCR signal in cattle compared with sheep as observed after experimental infections. It is noteworthy to mention that, in the vector-free period, much fewer sheep were sampled, whereas more healthy cattle were sampled to be moved or traded.
The PCR test described in the current study has also been used for research purposes to test many different types of samples, including organ suspensions, homogenates of ECE, and midges11,25 and ticks, 7 demonstrating the broad application of the PCR test in BTV research. The PCR test was further successfully used for detection of BTV serotype 6 in 2008. Discrimination between infection with BTV serotypes 8 and 6 would be possible by genetic differences in the regions between the conserved sequences of the primers and probe.22,23 At present, in the post-epizootic period, the PCR test is used to screen animals for international movement and monitoring as well as for research purposes.
In conclusion, a robust, thoroughly validated, high-throughput sample processing system using real-time RT-PCR detection for all BTV serotypes was described in the current study. The automated isolation of viral dsRNA combined with the all-in-one method and the 96-sample format systems can be used for development of BTV serotyping by PCR and for development of PCR tests for other serogroups of orbiviruses.
Footnotes
Acknowledgements
Peter Daniels/Ian Pritchard (CSIRO, Australia), Chris Hamblin, Carrie Batten, Chris Oura, and Peter Mertens (IAH-Pirbright, U.K.), Stephan Zientara (ANSES, France), and Samia Metwally (USDA, Plum Island, NY) are greatly acknowledged for providing virus strains. The KC cell line was kindly provided by Linda McHolland (USDA, Laramie, WY). The authors appreciated the sharing of unpublished sequences by Sushila Maan (IAH-Pirbright, U.K.), Martin Hofmann (IVI, Switzerland), and Christiaan Potgieter (Deltamune, South Africa). The authors also thank the animal caretakers for excellent assistance and Anoek Backx for veterinary supervision of the animal experiments. Many thanks to colleagues of CVI for testing field samples, particularly Yolanda de Visser-Hendrikse for recording positive and negative controls.
a.
ID VET Innovative Diagnostics, Montpellier, France.
b.
MagNA Pure isolation robot, version 4.05, Roche Diagnostics Nederland B.V., Almere, The Netherlands.
c.
Total Nucleic Acid Isolation Kit, Roche Diagnostics Nederland B.V., Almere, The Netherlands.
d.
LightCycler® RNA Master Hybridization Probes kit, Roche Diagnostics Nederland B.V., Almere, The Netherlands.
e.
RNA master SYBR Green I kit, Roche Diagnostics Nederland B.V., Almere, The Netherlands.
The author(s) declare that they do not have any conflict of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study has been funded by the Dutch Ministry of Economic Affairs, Agriculture, and Innovation as part of the agreement for statutory tasks.