
Abstract
The Reticuloendotheliosis virus (REV) group of retroviruses infects a wide range of avian species, including chickens, turkeys, ducks, geese, quail, and prairie chickens. The objective of the present study was to develop a highly sensitive and specific diagnostic test for the detection of REV in whole blood samples. In order to increase the diagnostic sensitivity, a duplex real-time polymerase chain reaction (PCR) that detects both the envelope protein gene (env) and the long terminal repeat (LTR) region of REV was designed. This assay demonstrated greater analytical and diagnostic sensitivity than the gel-based PCR assay when using DNA extracted from whole blood by both phenol-chloroform and magnetic bead methods. In general, threshold cycle values in the duplex real-time PCR assay were lower from DNA extracted using the magnetic bead system compared to DNA extracted by the phenol-chloroform method. Data presented herein show the successful development of a rapid and accurate test procedure, with high-throughput capability, for the diagnosis of REV infection using avian blood samples.
Reticuloendotheliosis virus (REV) is a type C retrovirus, and a member of the genus Gammaretrovirus within the family Retroviridae, subfamily Orthoretrovirinae. 16 The REV group is immunologically, morphologically, and structurally distinct from the leukosis/sarcoma group of avian retroviruses and currently consists of the following strains: nondefective REV-A, defective REV-T, Chick syncytial virus, Duck infectious anemia virus, and Trager duck spleen necrosis virus. 23 The host range of REV has been reported to include a variety of avian species, including chickens, turkeys, ducks, geese, quail, and prairie chickens.9,23 The most common disease syndromes associated with this group of viruses are a runting disease syndrome, chronic neoplasia of lymphoid and other organs, and acute reticulum cell neoplasia.8,11,23 Although infection seems to be widespread, the appearance of disease is uncommon and, in chickens, has been most frequently associated with REV-contaminated vaccines, such as Fowlpox virus (FPV) vaccines.10,22,23 Both remnants of the REV long terminal repeat (LTR) regions or near full-length REV provirus have been shown to be integrated into the genome of FPV and are implicated in virulence alteration.14,15,20 In 1998, REV was first diagnosed in the endangered Attwater’s prairie chicken (Tympanuchus cupido attwateri), and its presence in captive populations of this species continues to hamper recovery efforts.9,25
Various methods have been previously described to detect REV in avian blood and tissue samples, including virus isolation, immunohistochemistry, in situ hybridization, enzyme-linked immunosorbent assay, and fluorescent antibody test, but they can be time consuming (e.g., several weeks for virus isolation). 12 In comparison, molecular techniques such as polymerase chain reaction (PCR) have been used in the diagnosis of various infectious diseases and are considered one of the best alternatives to conventional assays because of their sensitivity, specificity, and reproducibility.15,17 Conventional gel-based PCR tests,5,6,10 as well as a nested PCR assay, 2 have been developed for routine REV diagnostics; however, these methods require manipulations post-PCR amplification including gel electrophoresis, gel staining steps, or transfer of amplified products to secondary tubes for further amplification. These methods can be time consuming and are prone to carryover contamination.
The objective of the present study was to develop a high-throughput process for the rapid diagnosis of REV with high sensitivity and specificity. To accomplish this, a duplex real-time PCR assay was designed to detect both the envelope protein gene (env) and the LTR regions of REV proviral DNA. In addition, 2 nucleic acid extraction methods were compared: a magnetic particle DNA extraction procedure and the commonly used phenol-chloroform extraction method.
For the real-time PCR assay described herein, primers and probes were designed using commercially available software a based on published sequences of the REV env gene and LTR sequences (GenBank accession nos. env: DQ513317, FJ439120, FJ439119, AF246698, DQ387450, and FJ496333; LTR: FJ439119, DQ387450, S70398, GQ870289, M22224, and GQ870290). All primers b and probes c were synthesized commercially, and the probes for env and LTR were labeled with fluorescence reporter dyes FAM and VIC, respectively. The following primers and probes were used in the duplex real-time PCR assay: env-F 5′-TCACTCTCGATGGAAATTG CAG-3′, env-R 5′-CCAGTCCTATTGTCTGCTTCCC-3′ (amplicon size 96 nt), and env-probe 6FAM-TAGATGT CAACTGCTATGCA-MGBNFQ; LTR-F 5′-AGGCTCAT AAACCATAAAAGGAAATGT-3′, LTR-R 5′-CCTTTACAA CCATTGGCTCAGTATG-3′ (amplicon size 119 nt), and LTR-probe VIC-ACAAACACGAGATCGAACTA-MGBNFQ.
Two DNA extraction methods were compared in the current study: 1) phenol-chloroform and 2) a microsphere paramagnetic bead kit d and processor. e The phenol-chloroform extraction was performed according to standard methods for the isolation of DNA. Briefly, 10 µl of whole blood was added to 500 µl of 1× STET buffer f (100 mM NaCl, 10 mM Tris–HCl [pH 8.0], 1 mM ethylenediamine tetra-acetic acid [pH 8.0], and 5% Triton X-100) and 20 µl of proteinase K g (20 mg/ml) and incubated for 15 min at 56°C. Then, 500 µl of buffered phenol-chloroform–isoamyl alcohol 25:24:1 f was added, and the sample was vortex mixed and then centrifuged for 5 min at 12,000 × g. The aqueous phase was collected, transferred to a new tube, and then 500 µl of chloroform–isoamyl alcohol (24:1) was added, vortexed, and centrifuged for 5 min at 12,000 × g. The aqueous phase was transferred to a new tube, and the DNA was precipitated by adding 100% ethanol (1:2, v:v) and 3 M sodium acetate, pH 5.2 (1:0.1, v:v), mixed by inverting several times, and incubated at −20°C for at least 2 hr. The DNA was pelleted by centrifugation at 12,000 × g for 5 min, air-dried, and resuspended in 90 µl of nuclease-free water. The DNA extraction procedure using the magnetic beads was performed according to manufacturer’s instructions d using a magnetic particle processor in a 96-well format. e Briefly, 10 µl of whole blood collected in heparin was added to 500 µl of lysis buffer, followed by the addition of 20 µl of bead mix, and incubated for 5 min at room temperature. The beads were then washed twice with wash buffer 1, twice with wash buffer 2, dried for 4 min, and purified DNA was eluted from the beads in 90 µl of nuclease-free water.
The duplex real-time PCR reactions were performed using a commercially available kit, h with 10 pmol of each env primer, 20 pmol of each LTR primer, 3 pmol of env probe, 6 pmol of LTR probe, and 3 µl of template DNA in a 25-µl final reaction volume. A no template control and a PCR amplification control (DNA from an REV-positive sample) were included as negative and positive controls, respectively. The PCR amplification and fluorescence detection were carried out in a real-time PCR thermocycler i and consisted of initial incubations at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 60 sec. Fluorescence was collected during the second step (annealing/extension) of the PCR process. Confirmation of the amplicons (both env and LTR) was determined by sequencing 2 independently cloned PCR products.j,k
Conventional gel-based PCR for detection of REV LTR was performed using a previously described method 6 with minor modifications. Briefly, the PCR reaction mixture consisted of 1.5 mM MgCl2, 200 µM of each deoxyribonucleotide triphosphate (dNTP), 1.25 U of Taq DNA polymerase, l 23.92 pmol of each primer, and 3 µl of template DNA in a final reaction volume of 25 µl. The amplification was carried out in an automated thermocycler m with the following parameters: initial incubation at 94°C for 5 min followed by 40 cycles of 94°C for 45 sec, 55°C for 45 sec, and 72°C for 45 sec, and a final elongation at 72°C for 10 min. The PCR amplification products were visualized by electrophoresis on 1.5% agarose gels using a fluorescent nucleic acid gel staining method. n
The specimens included in the present study consisted of 176 avian blood samples (168 Attwater’s prairie chickens, 6 chickens, 1 duck, and 1 pheasant) submitted to the Texas Veterinary Medical Diagnostic Laboratory (College Station, Texas) for various reasons, such as health checks or disease diagnostics, 27 of which had been confirmed REV positive and 149 negative by conventional gel-based PCR. Whole blood samples were stored at −20°C following initial screening. For the current study, blood samples were thawed and DNA extracted using both the phenol-chloroform and magnetic bead methods and tested by the newly developed duplex real-time PCR. Additionally, to test the specificity of the assay, DNA extracted from 9 Marek’s disease virus (MDV)-positive and 16 Avian leukosis virus (ALV)-positive field samples were included since MDV, ALV, and REV produce similar histological lesions. 23
The limit of detection of the duplex real-time PCR was determined using a plasmid containing the full-length REV proviral DNA diluted 10-fold serial dilutions (10−1–10−9). In addition, the detection limit of the duplex real-time PCR was also compared to the conventional gel-based PCR using DNA extracted from serial 10-fold dilutions (10−1–10−8) of an REV-positive whole blood sample. Plasmid containing REV proviral DNA was purified from bacterial culture using a plasmid mini prep kit, g and the DNA concentration was determined. o Results show that the limit of detection for the duplex assay was 46 and 9 copies for env and LTR, respectively (data not shown). The duplex real-time PCR was 10-fold more sensitive than the gel-based PCR with both the phenol-chloroform and magnetic bead extracted DNA (Table 1).
Comparison of the Reticuloendotheliosis virus (REV) duplex real-time polymerase chain reaction (PCR) assay to the conventional gel-based PCR method using serial dilutions of REV-positive avian blood and 2 nucleic acid extraction methods.*
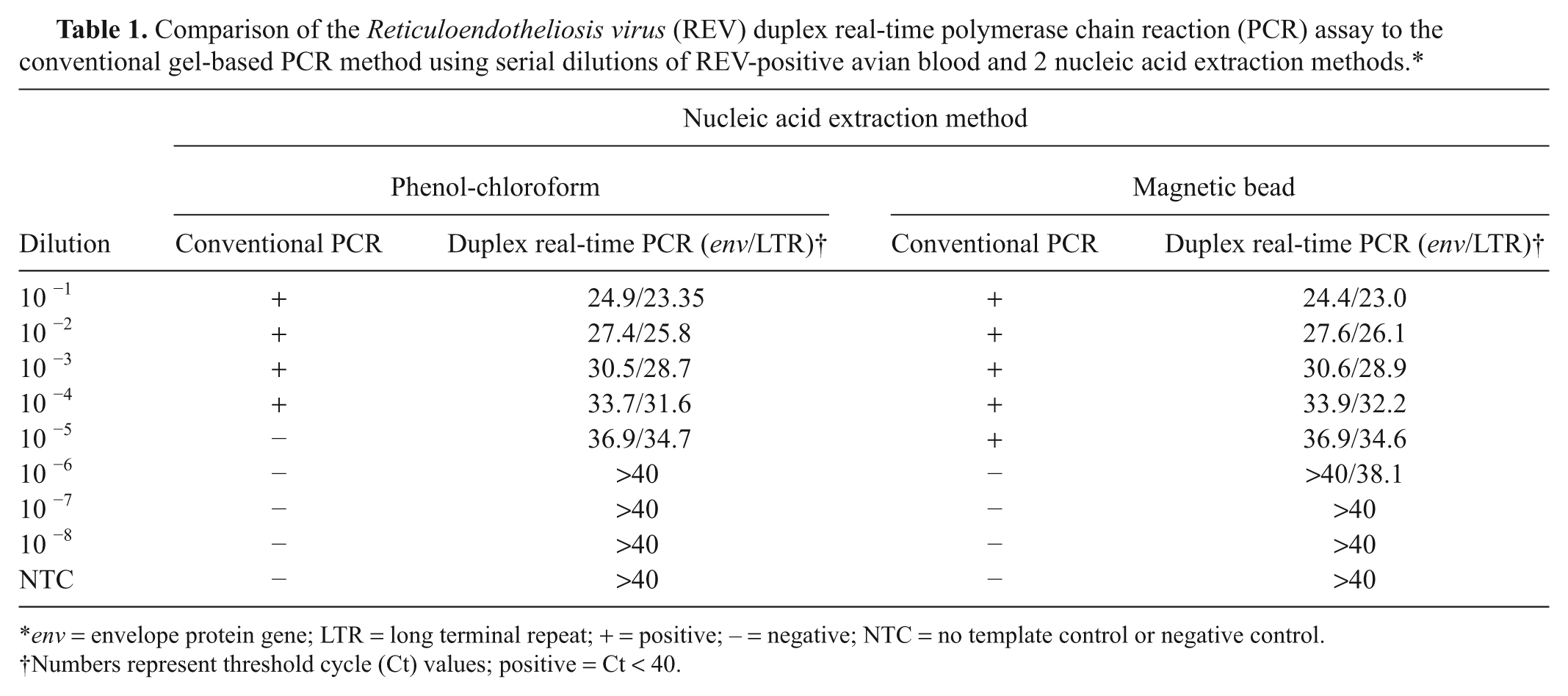
env = envelope protein gene; LTR = long terminal repeat; + = positive; – = negative; NTC = no template control or negative control.
Numbers represent threshold cycle (Ct) values; positive = Ct < 40.
The diagnostic sensitivity and specificity of the duplex real-time PCR assay was determined by comparison of DNA extracted from 176 avian blood samples previously screened for the presence of REV using the standard gel-based assay (27 REV positive, 149 REV negative). 6 Results show all 27 previously identified positive samples were positive for both env and LTR in the duplex real-time PCR when DNA was isolated using the magnetic bead extraction method (100% sensitivity [SE]). On the other hand, only 26 and 19 of the samples extracted using the phenol-chloroform method were positive for env (96.3% SE; 95% confidence interval [CI]: 79.1–99.8%) and LTR (70.4% SE; 95% CI: 49.7–85.5%), respectively, in the duplex real-time PCR assay. All previously REV-negative samples were also negative in the duplex assay with both extraction methods. In general, threshold cycle (Ct) values in the duplex assay were lower for both the LTR and env with nucleic acid extracted using the magnetic bead process compared to the phenol-chloroform method (Fig. 1). The duplex real-time PCR assay showed 100% specificity, as none of the previously identified negative samples nor the MDV- or ALV-positive samples were positive for REV (data not shown).
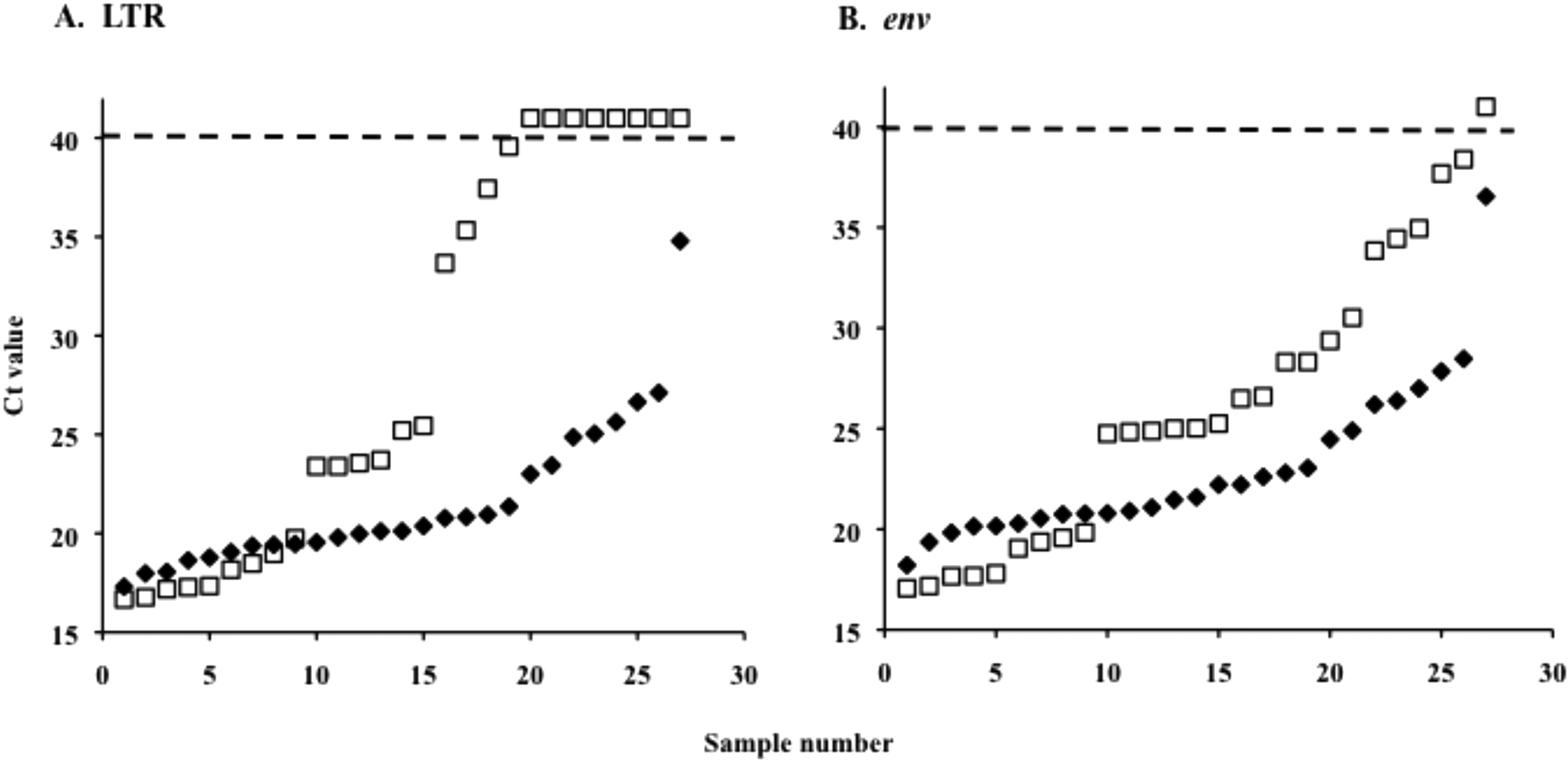
Comparison of 27 Reticuloendotheliosis virus (REV)-positive blood samples from Attwater’s prairie chickens extracted using 2 different methods and tested using the duplex REV real-time polymerase chain reaction (PCR) assay. Open squares represent DNA extracted by phenol-chloroform method, and solid diamonds represent DNA extracted by magnetic bead method. Dashed line depicts the threshold cycle value (Ct) above which samples were considered negative (Ct ≥ 40). env = envelope protein gene; LTR = long terminal repeat.
Decision makers rely on diagnostic tests to aid in management decisions regarding disease status. Reticuloendotheliosis virus has been a problem for the Attwater’s prairie chicken recovery efforts since it was first diagnosed in the captive breeding flock in 1995. 9 Due to the endangered status of this species, conventional control strategies employed in poultry operations, such as depopulation, cannot be applied. Thus, managers have to implement other control strategies, and a rapid, accurate test result is imperative to these efforts. Herein, a duplex real-time PCR assay that simultaneously detects 2 targets within the REV proviral genome is described.
Since REV is not a widespread problem in poultry and, when detected, terminal methods of control are implemented, a limitation in available REV-positive test specimens existed. The unique situation with the Attwater’s prairie chicken allows for the availability of REV-positive field samples from the captive flock. Although the majority (95.5%) of samples tested and all REV-positive samples were from Attwater’s prairie chickens, the assay could be applied to other species and specimens. Genomic sequences among REV isolates have been shown to have little variability, emphasizing the applicability of PCR-based tests for the detection of REV to various avian species.6,9,18 Additionally, the currently used assay for the detection of REV in clinical specimens was designed for use in poultry and is routinely used for other avian species, including the Attwater’s prairie chicken.
In conclusion, the use of real-time PCR increased the diagnostic sensitivity of the assay 10-fold compared to the traditional gel-based PCR assay, regardless of extraction method. Combining the magnetic bead extraction method and the real-time assay increased the diagnostic sensitivity 100-fold compared to the traditional phenol-chloroform extraction with the gel-based PCR assay. The ability of the duplex assay to detect fewer copies of LTR compared to env is not surprising in that the REV genome contains 2 copies of the LTR and only 1 copy of env. 1
Although not shown in the present study, the duplex real-time PCR assay described herein potentially has broader application since the REV proviral DNA is known to integrate in part (LTR only) or in whole into the genome of the infected host and large DNA viruses such as FPV and MDV.4,10,14,19,20,24 Thus far, the genomes of commercial vaccine strains of FPV and MDV field strains have been shown to contain complete or truncated REV-LTR, but not REV-env.4,19,21,24 On the other hand, field strains of FPV have been shown to have both REV-LTR and env.7,21 By incorporating the 2 targets, LTR and env, in the duplex assay, decision makers will be provided the ability to determine if the REV detected is potentially infectious or a partial insertion that would not result in a productive REV infection. Unfortunately, no samples were available for the current study that were positive for only one of the targets, LTR or env, thus further studies are needed to confirm this point.
The combination of nucleic acid isolation with magnetic beads and real-time PCR has been shown to greatly increase testing capacity of other avian pathogens, such as Newcastle disease virus and Avian influenza virus.3,13 Data presented herein show that the simultaneous use of a magnetic bead extraction method and a duplex real-time PCR assay provides a rapid and accurate test procedure for the detection of REV.
Footnotes
Acknowledgements
The authors wish to thank Jennifer Meier for her expert technical assistance. This work was a collaborative effort between the Texas Veterinary Medical Diagnostic Laboratory (TVMDL), the Texas A&M University College of Veterinary Medicine (TAMU-CVM), the U.S. Fish and Wildlife Service Attwater Prairie Chicken National Wildlife Refuge, and the Houston Zoo, Inc. The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views of the U.S. Fish and Wildlife Service.
a.
Primer Express™ 3.0, Applied Biosystems, Foster City, CA.
b.
Sigma Genosys, Sigma-Aldrich, St. Louis, MO.
c.
TaqMan®, Applied Biosystems, Foster City, CA.
d.
MagMAX 96 Viral RNA Isolation Kit, AM1836; Applied Biosystems, Foster City, CA.
e.
KingFisher 96, Thermo Scientific, Waltham, MA.
f.
Fisher Scientific, Pittsburg, PA.
g.
Qiagen Inc., Valencia, CA.
h.
FastStart Universal Probe Master (Rox), Roche Applied Science, Indianapolis, IN.
i.
ABI Prism 7000 and 7500 Fast Real-Time PCR detector, Applied Biosystems, Foster City, CA.
j.
TOPO-TA cloning® kit, Invitrogen Corp., Carlsbad, CA.
k.
BigDye Terminator v3.1 Cycle Sequencing Kit, ABI 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA.
l.
Promega Corp., Madison, WI.
m.
GeneAmp PCR System 9700, Perkin-Elmer, Waltham, MA.
n.
GelRed™, Phenix Research Products, Candler, NC.
o.
NanoDrop 8000, Thermo Scientific, Waltham, MA.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.