
Abstract
Due to the lipid rich cell wall of Mycobacterium avium subspecies paratuberculosis (MAP), the complex nature of bovine feces, and intermittent organism shedding by infected cattle, it is difficult to recover a sufficient amount of high-quality MAP DNA from fecal samples, directly affecting the sensitivity of downstream polymerase chain reaction (PCR) tests. In the current study, a DNA extraction method, designated the Mississippi Veterinary Research and Diagnostic Laboratory (MVRDL) method, was developed for PCR-based detection of MAP in bovine fecal samples. The MVRDL method combined multiple procedures, including chemical pretreatment, 1-tube cell lysis and extraction, chelex matrix absorption, and mini-column purification. The DNA yield and purity, as measured by spectrophotometry, was 3.36 fg per colony forming unit (CFU) MAP and A260/280 absorbance ratio of 2, respectively. This method was further evaluated by real-time PCR. A linear correlation was found between cycle-threshold (Ct) and log input CFU (ranging from 7.2 to 7.2 × 107 CFU per ml or CFU per g). The detection limit of the real-time PCR assay was 3 CFU per ml of MAP culture or per g of MAP-spiked feces. In addition, the MVRDL method was validated by performing 7 Johne's direct fecal PCR proficiency tests administered by the National Veterinary Service Laboratories. Based on culture results as the “gold standard,” the specificity of MVRDL PCR was 100%, and the sensitivity was 98.46% for samples containing more than 1.5 CFU per tube of fecal cultures. To the authors' knowledge, this is the most efficient MAP DNA extraction method in comparison with all previously published protocols.
Introduction
Johne's disease, also known as paratuberculosis, is prevalent worldwide and causes a significant economic loss to the cattle industries. 17,23 The disease manifests as a chronic, progressive enteritis due to infection with Mycobacterium avium subspecies paratuberculosis (MAP). 4 Excretion of the organism may occur intermittently for a prolonged period before the onset of clinical disease. 23 Current strategies for control and eradication of Johne's disease in many countries include surveillance of herds for infected animals, culling, and improving herd management. 4,17 The disease monitoring and diagnosis rely heavily on laboratory testing, including enzyme-linked immunosorbent assay (ELISA) detection of antibody production, bacteriologic culture, and molecular identification of MAP genetic materials. 5,6,23 The prolonged subclinical stage, accompanied by varying magnitudes of immune response and organism shedding, as well as the slow-growing nature of MAP make the disease diagnosis a complicated task. 9,22,23
During the past decade, numerous polymerase chain reaction (PCR)-based tests have been used in confirming serological and bacteriological results, as in the case of radiometric culture in liquid media. 2,8,10,13 To develop highly sensitive and specific molecular detection methods, research has been focused on finding target sequences that are specific to MAP and abundant for detection, choosing suitable equipments and optimal PCR and/or hybridization conditions, evaluating DNA extraction methods, and optimizing techniques for recovering MAP organisms from clinical specimens. 2,9,11,14,17 The insertion sequence IS900, a 1451 base-pair (bp) repetitive element present in 12–20 copies in MAP genome, is commonly targeted for the detection of MAP by PCR. 2,10 Other sequences, such as hspX and F57, have also served as amplification targets for MAP PCR. 8,11 Because of the thick and lipid rich cell wall, it is difficult to release genetic materials from MAP. 1 To overcome this difficulty, chemical lysis and physical disruptions, with and without subsequent purifications, have been used by various groups. 3,21,22 Despite the enormous amount of effort, a highly sensitive, specific, and cost-effective method for early diagnosis of Johne's disease remains to be developed. 22,23 In an attempt to achieve such a goal, a DNA extraction method for PCR detection of MAP organisms in bovine fecal samples was developed, the efficiency of which was determined by spectrophotometry and TaqMan real-time PCR.
Materials and methods
Bacterial strain and growth conditions
The MAP strain American Type Culture Collection (ATCC) 19698 was cultured in 7H9 broth a supplemented with 2.5% (vol/vol) glycerol, b oleic acid-albumin-dextrose-catalase, a 0.05% Tween 80, a and 2 mg of mycobactin J, c as described previously. 11 The purity of the MAP cultures was periodically confirmed by staining with the Ziehl–Neelsen acid-fast method and inoculating tryptic soy agar (TSA) with 5% sheep's blood. a To determine the number of colony forming units (CFU), serial dilutions of MAP cultures (100 μl) were inoculated onto Herrold egg yolk (HEY) slants with mycobactin J a and incubated at 37°C for 16 weeks. To culture MAP organisms from feces, specimens were processed as previously described. 19 In brief, 1 gram of feces was thoroughly mixed with 35 ml of double-distilled water for 30 min and allowed to stand undisturbed at room temperature for 30 min. The supernatant (30 ml) was transferred to a new tube and centrifuged at 1,800 × g for 30 min. The pellet was resuspended in 30 ml of HPC-BHI (0.9% cetylpyridinium chloride and 1.9% brain heart infusion), incubated overnight at 37°C, and centrifuged again. The resultant pellet was resuspended in 1 ml of antibiotic brew (1.9% BHI broth containing 50 μg of amphotericin B/ml, 100 μg of vancomycin/ml, and 100 μg of nalidixic acid/ml). Following another overnight incubation at 37°C, 200 μl of the suspension was inoculated onto 4 HEY slants with mycobactin J and 1 HEY slant without mycobactin J as negative control. The inoculated slants were incubated for 16 weeks at 37°C. At the end of incubation, colonies that appeared on slants with mycobactin J, but not the mycobactin J–negative control, were stained by the Ziehl–Neelsen method and/or subjected to PCR for confirmation.
Fecal sample collection
Equal amounts of fecal material from five 4-year-old Angus cows with multiple negative fecal cultures were pooled, mixed thoroughly, divided into 1-g aliquots, and stored at −80°C prior to being spiked with MAP. A total of 179 bovine fecal samples derived from 7 Johne's disease fecal proficiency tests administered by the National Veterinary Services Laboratory (NVSL) were used in validation studies. Each of these test kits contained 25 unknown samples and 1 known positive, except for the 2004 kit, which did not have a positive control. Two samples of low-shedding status in the 2008 proficiency test kit were eliminated from the test panel by NVSL because they were classified as negative by the majority of the participating laboratories.
Preparation of samples for DNA extraction
Mycobacterium avium subspecies paratuberculosis organisms from multiple exponential-phase (OD550 0.4–0.6) cultures were harvested by centrifugation at 1,800 × g for 30 min. The bacterial pellets were washed twice with 25 ml phosphate buffered saline (PBS) b supplemented with 0.25% Tween 80 b and resuspended in 5 ml PBS with 0.25% Tween 80. Single-cell suspensions were obtained by vortexing the samples for 20 sec followed by sonication for 3 min in an ultrasonic cleaner FS20D. d The number of bacteria in each suspension was estimated based on direct microscopic counts and then confirmed by CFU enumeration on HEY slants. Only the CFU counts were used in subsequent assessment of DNA yield and real-time PCR sensitivity. The bacterial concentration was adjusted to approximately 109 MAP organisms per ml based on the initial microscopic counts. Ten-fold serial dilutions ranging from 108 to 10 MAP organisms per ml were prepared in PBS supplemented with 0.25% Tween 80. b To prevent clumping, MAP suspensions were vortexed for 20 sec between dilution steps. Aliquots (100 μl) of MAP suspension and dilutions were mixed with the fecal aliquots (1 g) to yield samples containing 108 to 1 MAP organisms. The remaining bacterial suspensions were stored at −80°C for later use. Negative fecal controls were prepared by adding 100 μl PBS with 0.25% Tween 80 to each of the fecal aliquots used.
DNA extraction
Guanidine-lysis method. A 1-tube guanidine-lysis method 11 was modified to extract DNA from pure MAP culture and MAP-spiked fecal material. The MAP suspensions (1 ml) containing approximately 1–108 bacteria per sample were centrifuged at 13,000 × g for 10 min at 4°C. The supernatant was discarded, and the cell pellet was resuspended in 800 μl lysis buffer containing 0.01 M Tris-hydrochloric acid (HCl), b 0.005 M ethylenediamine tetraacetic acid (EDTA), b 4 M guanidine thiocyanate, b 0.25% sodium dodecyl sulfate (SDS), b and 0.5% sodium citrate. b The suspension was transferred into a fresh 2-ml tube containing 400 μl Tris-saturated phenol, b 400 μl chloroform, b and 0.5 g 0.1-mm-diameter zirconium-silica beads. e The tube was shaken in a bead-beater f at speed 5 for 45 sec, cooled on ice, and centrifuged at 8,000 × g for 5 min. The supernatant (500 μl) was mixed with 500 μl of Chelex100 matrix g and incubated at 56°C for 30 min and 100°C for 10 min. Following incubation, the mixture was centrifuged at 13,000 × g for 10 min at 4°C. The supernatant (800 μl) was mixed with 400 μl of 100% ethanol, passed through a mini spin column fitted with a silica membrane, h washed twice with 700 μl wash buffer (2 mM Tris-HCl pH 7.5, 150 mM sodium chloride [NaCl] in 80% ethanol), and eluted with 50 μl DNase- and RNase-free water. To extract DNA from fecal samples, each prepared fecal aliquot was thoroughly mixed with 25 ml 0.02 N sodium hydroxide (NaOH) and allowed to stand undisturbed at room temperature for 30 min. The supernatant (20 ml) was transferred to a new tube and centrifuged at 1,800 × g for 30 min at 25°C. The pellet resulting from the centrifugation of the supernatant was washed twice with 25 ml distilled water and centrifuged at 1,800 × g for 30 min at 25°C. The pellet was resuspended in 800 μl lysis buffer and subjected to DNA extraction as described above. This DNA extraction procedure was designated as the Mississippi Veterinary Research and Diagnostic Laboratory (MVRDL) MAP extraction method.
Kit A method. A commercially available DNA extraction kit i manufactured for general laboratory purpose was used to extract DNA from serially diluted MAP culture. A bead-beating step identical to that for the MVRDL method was incorporated into the protocol provided by the manufacturer. To fill the 2-ml bead beater tube, the volume of the sample resuspension buffer was doubled. Thereafter, the volumes of other reagents were adjusted proportionally.
Kit B method. Kit B, j another commercially available DNA extraction kit, was used to extract DNA from serially diluted MAP culture as well as MAP-spiked fecal samples. Since this kit was developed specifically for MAP DNA extraction, all procedures were performed strictly according to the manufacturer's instructions. With all 3 DNA purification methods, the final volume of DNA was 50 μl.
Determination of DNA yield and purity
The DNA concentration (μg/ml) derived from pure MAP culture was determined spectrophotometrically and calculated using the following formula: the absorbance at 260 nm (A260) × dilution factor × 50 × μg/ml. The DNA yield was calculated as follows: concentration × volume (50 μl)/number of bacteria (CFU) used for DNA extraction. The purity of DNA was expressed as the ratio of absorbance at 260 nm and 280 nm (A260/A280).
Real-time PCR assays
Quantitative real-time IS900-based PCR assays were performed in 25-μl volumes using a forward primer SF214 (5′-ATGACGGTTACGGAGGTGGTT-3′), a reverse primer SR289 (5′-TGCAGTAAT GGTCGGCCTTAC-3′), and a probe PR265 (5′-FAMCGACCACGC CCGCCCAGATAMRA-3′) as described previously with modifications. 19 In brief, the reaction mixture consisted of DNase- and RNase-free water, 1× TaqMan Universal PCR Master Mix, k 300 nM primers, 75 nM probe, and 5 μl of template DNA. The PCR primers and probe were commercially synthesized, l and the PCR assays were performed on a sequence detection system m under the following conditions: 1 cycle of 94°C for 5 min; 45 cycles of 95°C for 15 sec and 60°C for 1 min. A positive reaction control containing 10 fg MAP genomic DNA in 5 μl of water and a negative reaction control using 5 μl of sterile water to replace template DNA were included in each experiment. In addition, a commercial Johne's PCR kit (C) n was used to evaluate the efficiency of DNA extraction methods.
To minimize laboratory contamination, sample processing, DNA extraction, and PCR amplification were carried out in 3 separate rooms. Critical procedures, such as DNA extraction and reaction assembly, were performed in laminar-flow hoods. Workbenches and hoods were disinfected with 70% ethanol daily and examined periodically for contamination with MAP or other microbes by performing PCR tests and/or bacterial cultures.
Evaluation of the MVRDL MAP DNA extraction method
The MVRDL method was evaluated based on the following features: 1) the quantity and quality of DNA extracted from pure MAP culture, 2) the quantitative results of downstream real-time PCR, 3) the detection limits of real-time PCR assay for pure MAP culture and MAP-spiked fecal samples, 4) the overall performance of real-time PCR in multiple Johne's proficiency tests, and 5) the quantitative results (Ct values) of real-time PCR in 2 recent proficiency tests.
To achieve the first goal, DNA samples extracted from the same number of bacteria (3.6 × 108 CFU) using the 3 extraction methods were analyzed spectrophotometrically. The yields and purities were compared among extraction methods. Because DNA samples derived from MAP-spiked feces could contain DNA molecules originating from other fecal microorganisms or even the hosts, no attempt was made to quantify MAP DNA in these samples. To accomplish the second goal, DNA samples prepared from 10-fold dilutions of MAP culture (7.2 × 104 to 7.2 × 107 CFU per ml) were subjected to real-time PCR analysis. The Ct values were compared among extraction methods.
To determine the analytical sensitivity of the MVRDL real-time PCR, standard curves were generated by plotting the Ct values against log input CFU of MAP organisms used for DNA extraction. A cutoff Ct value was chosen to achieve maximum accuracy and minimize false-negative results. Then, the overall performance of the MVRDL MAP extraction real-time PCR was evaluated based on the agreement between real-time PCR and NVSL culture results. Finally, the sensitivity of the MVRDL MAP extraction real-time PCR method was compared with that of a commercial MAP extraction PCR system based on the Ct values for 50 fecal samples in 2 proficiency test kits.
Statistical analysis
The efficiencies of the 3 DNA extraction methods were compared using the data analysis package included within Microsoft Excel 2000 software. o Differences between methods were evaluated by a 2-tailed Student's t-test.
Results
Efficiency of the MVRDL DNA extraction method
The efficiency of the MVRDL MAP DNA extraction method was initially evaluated using pure MAP culture. The yield of DNA obtained with the MVRDL method (3.36 fg per CFU) exceeded that obtained by using kit A (1.88 fg per CFU) or kit B (1.04 fg per CFU). The purity of DNA prepared using the MVRDL method was consistently high, as indicated by the A260/A280 ratio of 2.0 (Fig. 1). In contrast, protein contamination was detected in DNA extracted by kit A (A260/A280 = 1.53) or kit B (A260/A280 = 1.08). When DNA samples prepared from pure MAP culture by the 3 extraction methods were subjected to real-time PCR analysis, Ct values for the MVRDL extraction method were significantly lower than for kit A or kit B (Fig. 2). The differences in Ct values between the MVRDL method and kit A were within 3 cycles, whereas the differences between the MVRDL method and kit B ranged from 3 to 8 cycles (Fig. 2). To rule out possible artifacts resulting from an incompatibility between DNA extracted by kit B and the in-house PCR reagents, additional real-time PCR assays were carried out using PCR reagents in kit C. The same manufacturer j , n developed kits B and C for MAP DNA extraction and PCR detection of MAP organisms in bovine fecal specimens. Following the replacement of in-house PCR reagents with kit C, slight reductions in real-time PCR Ct values were observed (Fig. 2). However, the Ct values obtained using the MVRDL extraction real-time PCR method were still significantly lower than that achieved by using kit B in conjunction with kit C. These data suggested that the MVRDL method was indeed more efficient than any of the commercial kit methods tested in the study.
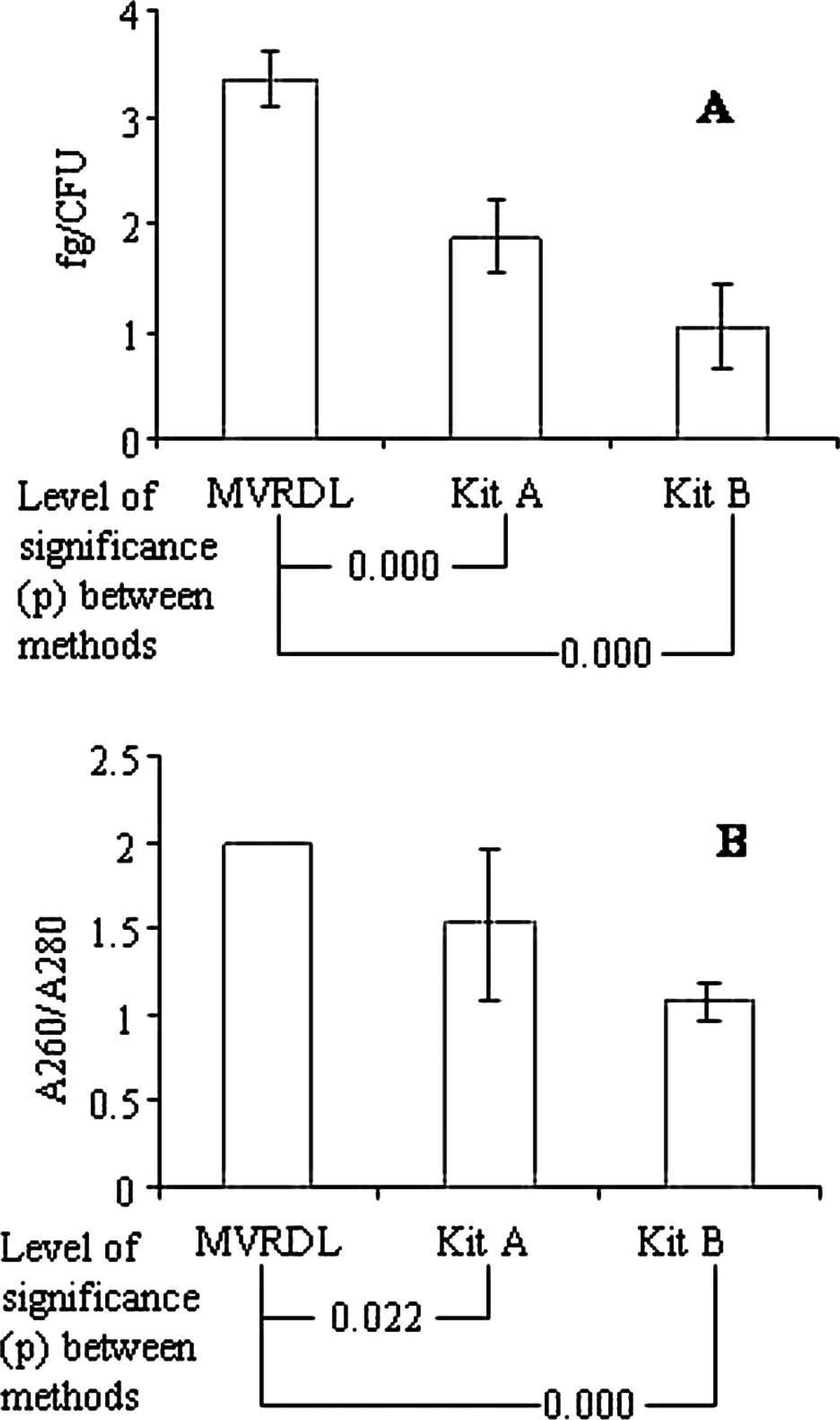
Quantitative (
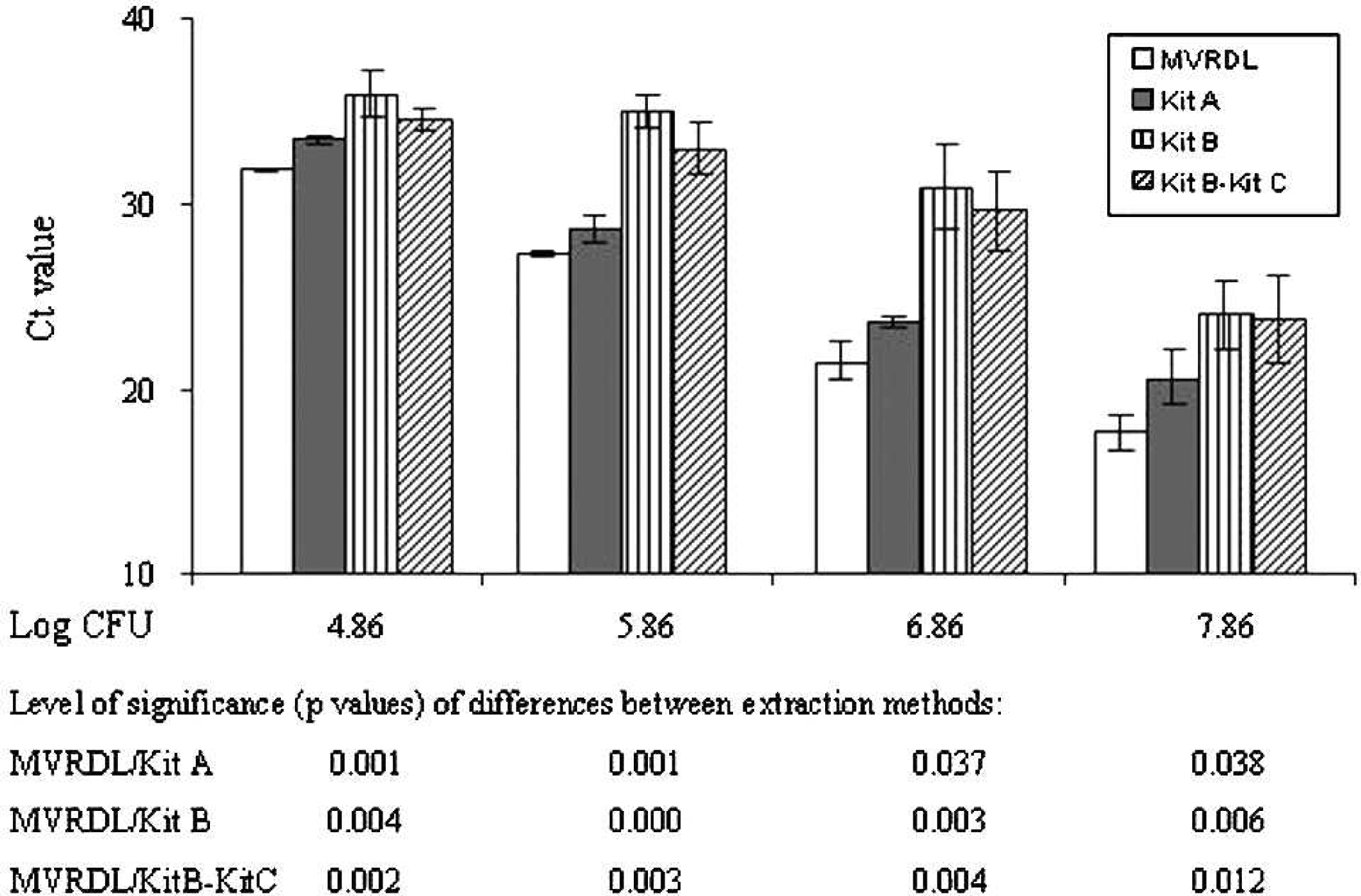
Efficiencies of the Mississippi Veterinary Research and Diagnostic Laboratory extraction method (MVRDL), kit A, h and kit B i as measured by quantitative real-time polymerase chain reaction (PCR). The DNA templates extracted from 10-fold dilutions of pure Mycobacterium avium subspecies paratuberculosis culture were evaluated using in-house PCR reagents. The DNA prepared with kit B i was also analyzed using commercial Johne's PCR reagents from the kit C. m Significances (P values) of differences between extraction methods are indicated below the bar graph. Data shown are average cycle threshold (Ct) values ± standard deviation of 3 independent experiments.
Analytical sensitivity of the MVRDL real-time PCR
Following the initial evaluations, the analytical sensitivity of MVRDL MAP real-time PCR was determined by using serial 10-fold dilutions of pure MAP culture or MAP-spiked fecal samples. A linear correlation was found between the Ct values ranging from 18.43 to 39.74 and the log input CFU ranging from 7.2 × 107 to 7.2 CFU per ml of culture or CFU per g of feces. The equation of Ct versus log CFU for pure MAP culture was Y = −3.2138x + 43.546 with an R 2 (coefficient of determination) of 0.9801 and an E (reaction efficiency) of 1.047. The equation of Ct versus log CFU for MAP-spiked fecal material was Y = −2.9402x + 43.541 with an R 2 of 0.9793 and an E of 1.188 (Fig. 3). When the number of input bacteria fell below 7.2 and above 3 CFU per ml (g), RT-PCR reactions were still positive and Ct values varied between 40 and 42. Based on the linear equations, the analytical sensitivity (or detection limit), with a Ct of 42 as the cutoff point, was 3.03 CFU per ml for pure MAP culture, or 3.34 CFU per g of MAP-spiked fecal samples. Reactions with Ct values between 42.01 and 44.99, which could theoretically detect between 3.01 and 0.36 CFU per ml of MAP culture or between 3.32 and 0.32 CFU per g of fecal sample, were treated as suspect. No amplification signals were obtained with the negative reaction controls (no template) or the negative fecal sample controls (samples spiked with sterile PBS). For the convenience of calculation, the maximum cycle number (45) was assigned to all reactions that failed to amplify the target gene.
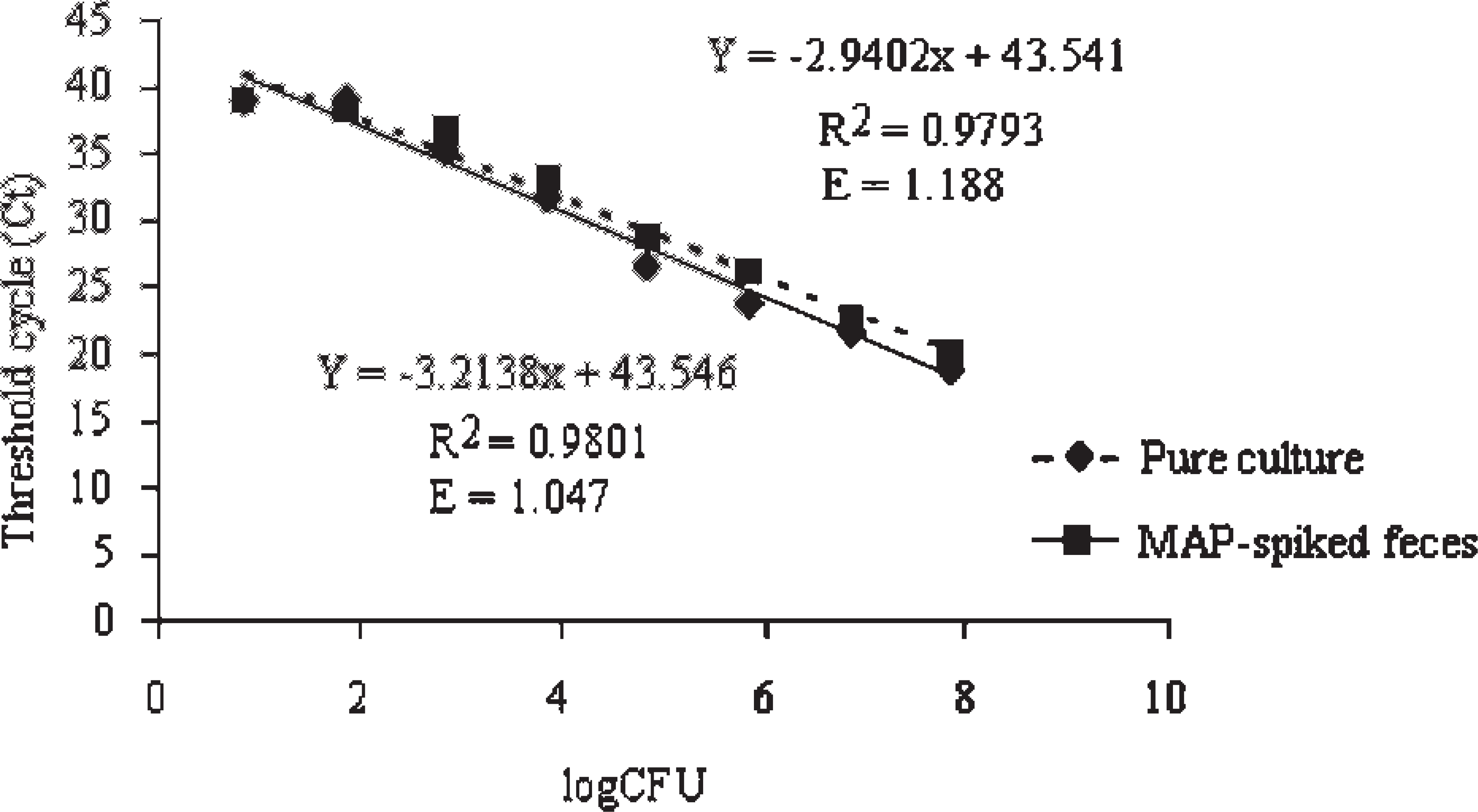
The detection limit and amplification efficiency of Mississippi Veterinary Research and Diagnostic Laboratory Mycobacterium avium subspecies paratuberculosis (MAP) extraction polymerase chain reaction. The lines represent the regression of log input colony-forming unit (CFU) of MAP in 1 ml of culture (solid line) and in 1 g of feces (dashed line). R 2 (the coefficient of determination) and E (the reaction efficiency) are indicated on the graph. Threshold cycle values are the average of 3 independent experiments.
Overall performance of the MVRDL real-time PCR on NVSL Johne's proficiency test
When the MVRDL MAP DNA extraction and real-time PCR method was used to complete 7 Johne's Disease Proficiency tests, all 49 negative samples were identified as negative and 128 out of 130 positive samples were identified as positive (Table 1). According to the shedding status provided by NVSL upon completion of the proficiency tests, the lowest MAP concentration in the proficiency test samples was 1.5 CFU per tube. Using fecal culture as a reference test, the MVRDL MAP real-time PCR showed a diagnostic specificity of 100% and a sensitivity of 98.46% for fecal specimens containing more than 1.5 CFU per tube.
Performance of the Mississippi Veterinary Research and Diagnostic Laboratory Mycobacterium avium subspecies paratuberculosis extraction polymerase chain reaction on National Veterinary Services Laboratory Johne's disease proficiency tests. *
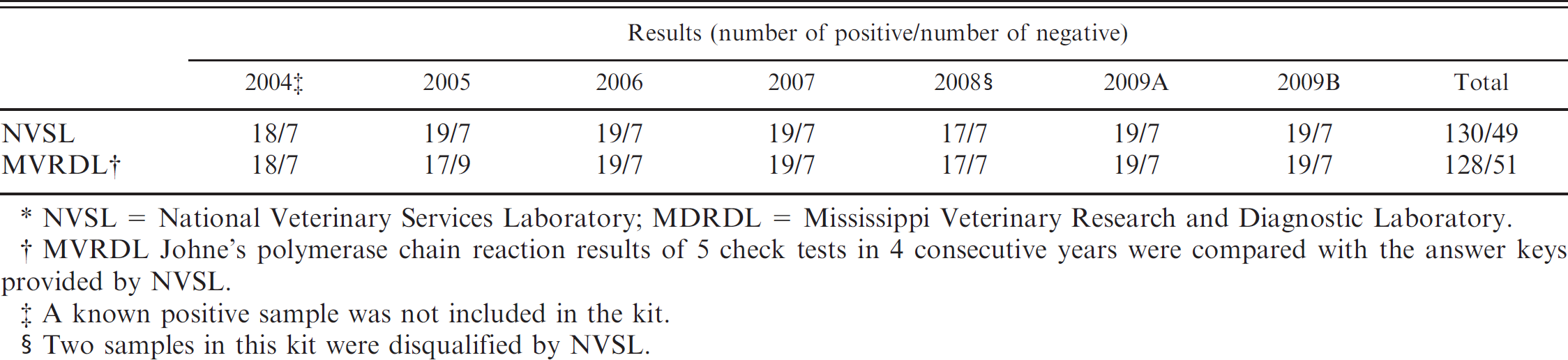
NVSL = National Veterinary Services Laboratory; MDRDL = Mississippi Veterinary Research and Diagnostic Laboratory.
MVRDL Johne's polymerase chain reaction results of 5 check tests in 4 consecutive years were compared with the answer keys provided by NVSL.
A known positive sample was not included in the kit.
Two samples in this kit were disqualified by NVSL.
Quantitative performance comparison of the MVRDL real-time PCR and a commercial MAP detection system
The MVRDL MAP DNA extraction real-time PCR and a commercial MAP extraction real-time PCR system (kit B/kit C) j , n were used to test 50 fecal samples derived from 2 proficiency test kits received in 2 consecutive years (2008 and 2009). A comparison of the quantitative results (Ct values) showed that the MVRDL method was more effective than the commercial system in detecting low numbers of MAP bacteria in bovine feces. The quantitative results of positive samples were summarized and are presented in Table 2. With the 12 low-shedding samples (1.5–6 CFU per tube), 3 samples (08-21, 08-25, and 08-07) from the 2008 proficiency test and 2 samples (09-21 and 09-24) from the 2009 proficiency test were detected by the MVRDL method but were missed by the commercial system (kit B and kit C). For the convenience of calculation, the maximum PCR cycle number (45) was assigned to these negative reactions. With the modest to high shedding samples, lower Ct values were frequently associated with the MVRDL method than with kit B and kit C. Inter-well and inter-sample variations for a few sets of replica samples (e.g., 09-15 and 09-23) were seen with samples belonging to different shedding categories and with both test methods. Thus, measurement or pipetting errors, instead of deficiencies associated with a given method, were more likely the cause of these variations. Finally, all 14 culture-negative samples were identified as negative (no amplification signals) by both DNA extraction real-time PCR systems, suggesting that both tests were highly specific.
Quantitative results of Mississippi Veterinary Research and Diagnostic Laboratory real-time polymerase chain reaction and a commercial direct fecal polymerase chain reaction. *
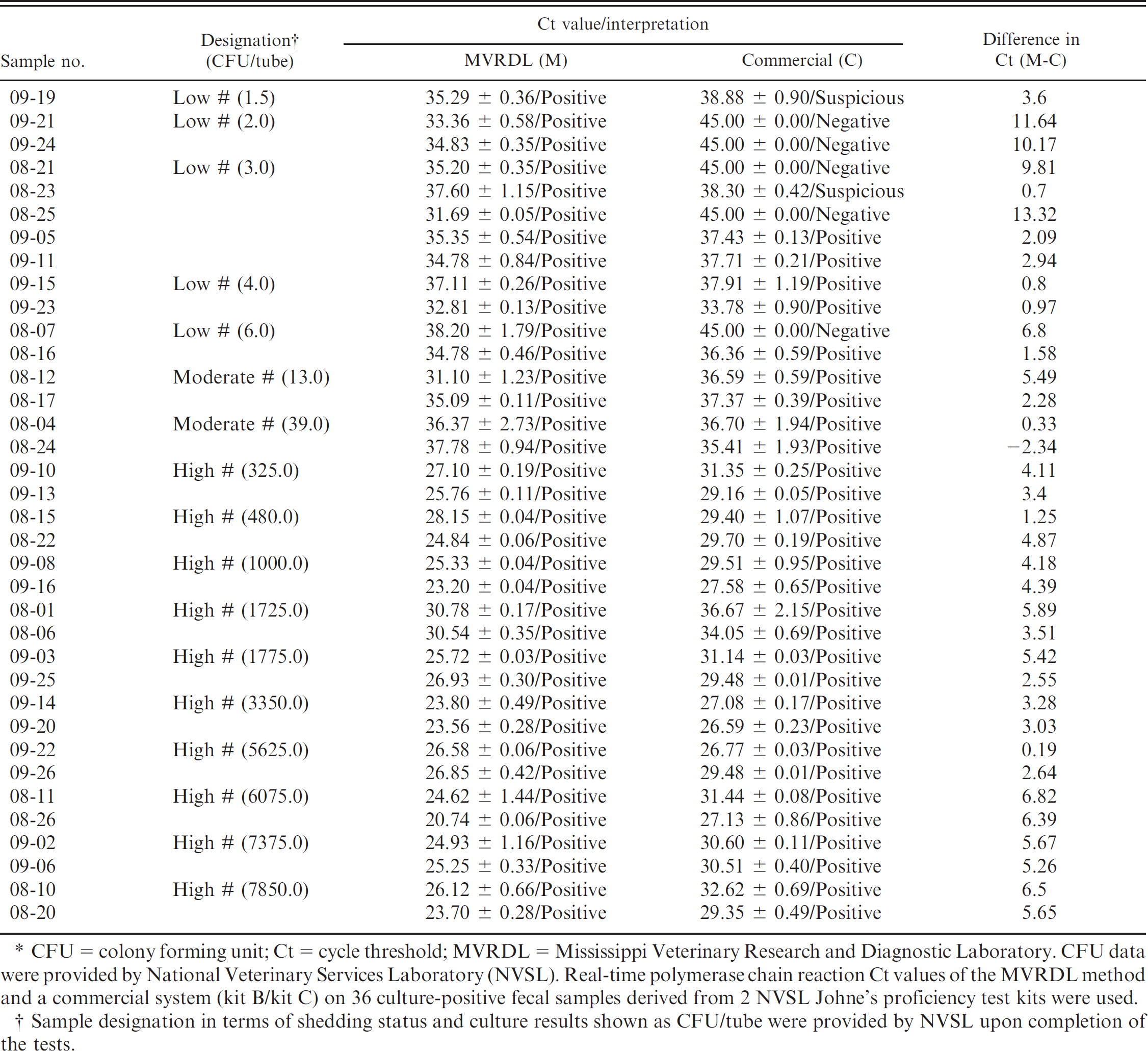
CFU = colony forming unit; Ct = cycle threshold; MVRDL = Mississippi Veterinary Research and Diagnostic Laboratory. CFU data were providedby National Veterinary Services Laboratory (NVSL). Real-time polymerase chain reaction Ct values of the MVRDL method and a commercial system (kit B/kit C) on 36 culture-positive fecal samples derived from 2 NVSL Johne's proficiency test kits were used.
Sample designation in terms of shedding status and culture results shown as CFU/tube were provided by NVSL upon completion of the tests.
Discussion
Although PCR assays aimed at detecting MAP organisms in liquid or solid cultures have been optimized, 1,3,16 direct fecal PCR tests remain to be improved to achieve high specificity and sensitivity. 5,9 A challenging task in the development of Johne's direct fecal PCR is obtaining a sufficient amount of inhibitor-free MAP DNA from clinical specimens. To date, various in-house protocols and commercial kits have been used to purify MAP DNA. 1,3,7,12,16 While all of them are useful, each has its own inherent limitations in terms of DNA yield and purity, the specificity and sensitivity of down-stream PCR, cost-effectiveness, and practicability. The current study aimed to develop an efficient and cost-effective DNA extraction method for PCR-based detection of MAP organisms in bovine feces. The method used combined multiple procedures, including NaOH pretreatment, 1-tube cell lysis and extraction, chelex resin absorption, and mini-column purification. The study did not focus on the development of a PCR procedure, because highly specific and efficient PCR primers and amplification conditions had been reported previously. 19
The guanidine lysis method has been used to extract mycobacterial DNA in human specimens. 14 The application of this method in Johne's diagnosis is limited due largely to its inefficiency in removing PCR inhibitors originating from bovine fecal specimens. 3 In the current study, additional procedures were incorporated into the traditional guanidine lysis method to make it more suitable for the purification of MAP DNA from bovine fecal samples. First, NaOH pretreatment facilitated the liberation of MAP organisms bound within fecal matter and/or contaminated soil materials, thereby improving the process of organism recovery 19 ; the step also helped to eliminate PCR inhibitors as reported previously. 20 The combination of mechanical disruption and 1-tube chemical lysis/extraction allowed for maximal recovery of MAP DNA and reduction of cross-contamination. 14 The chelex matrix absorbed metal ions that could catalyze DNA degradation and inhibit downstream PCR reaction. 15 Finally, a mini-column purification procedure quickly removed the contaminating carbohydrates. Although multiple steps were involved, the MVRDL MAP extraction procedure could be completed in about 3 hr, and the cost for reagents was less than $3 per sample.
During the initial stage of method development, the MVRDL protocol was compared with 2 commercial DNA extraction kits, one for general DNA isolation and the other for MAP DNA extraction. These comparisons were performed using serial dilutions of pure MAP culture. The data clearly indicated that the MVRDL method was more efficient than the 2 commercial kits in terms of DNA yield and purity as well as the Ct values of downstream RT-PCR assay. The DNA purity (A260/A280 = 2) obtained using the MVRDL method was also much higher than what had been reported by other groups (A260/A280 = 1.3–1.7). 1,12 The effectiveness of this DNA extraction method enabled the downstream real-time PCR to achieve a remarkably high analytical sensitivity, approximately 3 CFU per ml culture (or per g feces).
Validation of the MVRDL method was conducted by performing “blind” tests on fecal samples derived from multiple Johne's proficiency test kits. The MVRDL real-time PCR demonstrated a specificity of 100% and a sensitivity of 98.49%. These values greatly exceeded the previously reported specificities (51.5–99%) and sensitivities (70.2–85.3%) of several Johne's PCR tests. 5,9,22 The accuracy and consistency of the MVRDL MAP real-time PCR on multiple Johne's proficiency tests proved that this method could be widely adopted for the diagnosis of Johne's disease. Taken together, data from the study showed that the MVRDL MAP extraction method is an efficient and inexpensive alternative to the commercially available MAP DNA extraction reagents. The high purity and yield of the DNA obtained suggest that the method can be potentially applied to extract DNA for molecular detection of other bacterial pathogens in bovine feces.
Acknowledgements
The study was partially funded by the Mississippi Board of Animal Health. The authors thank Dr. Carla Huston for providing bovine fecal samples; Dr. Sue Ann Hubbard for critical review of the manuscript; and Ms. Candy Zhang, Mr. Anthony Liu, and Mr. Gabriel Senties for technical assistance.
Footnotes
a.
Becton, Dickinson and Company, Sparks, MD.
b.
Sigma-Aldrich, St. Louis, MO.
c.
Allied Monitor, Inc., Fayette, MO.
d.
Thermo Fisher Scientific, Waltham, MA.
e.
BioSpec Products, Bartlesville, OK.
f.
FastPrep® FP120 Cell Disrupter, Qbiogene Inc., Carlsbad, CA.
g.
Bio-Rad Laboratories, Hercules, CA.
h.
Epoch Biolab, Sugarland, TX.
i.
QIAamp DNA Mini Kit, Qiagen Inc., Valencia, CA.
j.
MAP Extraction kit, Tetracore Inc., Rockville, MD.
k.
Applied Biosystems, Foster City, CA.
l.
Eurofins MWG Operon, Huntsville, AL.
m.
ABI Prism® 7000, Applied Biosystems, Foster City, CA.
n.
VetAlet™ Johne's Real-Time PCR kit, Tetracore Inc., Rock-ville, MD.
o.
Microsoft Corp., Redmond, WA.