
Abstract
Salmonella enterica is an important enteric pathogen consisting of many serovars that can cause severe clinical diseases in animals and humans. Rapid identification of Salmonella isolates is especially important for epidemiologic monitoring and controlling outbreaks of disease. Although immunologic and DNA-based serovar identification methods are available for rapid identification of isolates, they are time consuming or costly or both. In the current study, 2 molecular methods for identification of Salmonella serovars were developed and validated. A 70-mer oligonucleotide spotted microarray was developed that consisted of probes that detected genes responsible for genetic variation among isolates of Salmonella that can be used for serotyping. A multiplex polymerase chain reaction (PCR) assay was also developed, which is capable of identifying 42 serovars, thus providing a valuable prediction of the pathogenicity of the isolates by detecting the presence of virulence genes sseL, invA, and spvC. The gene spvC was the best predictor of pathogenicity. In a blind study, traditional serologic methods were correlated at 93.3% with the microarray-based method and 100% with the multiplex PCR-based serovar determination.
Keywords
Introduction
Gastrointestinal tracts of both humans and animals are a major habitat for Salmonella. In 2005, more than 36,000 clinical cases of salmonellosis were reported to the Centers for Disease Control and Prevention. 10 Of these, 15,000 resulted in hospitalization, and 400 were fatal. 22 Salmonella enterica subspecies enterica, with most clinical cases belonging to subgroup I, is categorized into serogroups (based on lipopolysaccharide or O antigens) and serotypes (based on the flagellar or H antigens). 40
To identify the individual serovars (based on O and H antibody-antigen tests) of Salmonella, testing is routinely performed in state reference laboratories and in several veterinary laboratories, which, because of the high number of samples submitted, can lead to long turnaround times. In an attempt to streamline the process, molecular techniques have been developed, such as ribotyping, 14 polymerase chain reaction (PCR) single-strand conformation polymorphism analysis, 34 restriction fragment length polymor-phism, 23 pulsed-field gel electrophoresis, 4,25 IS200 fingerprinting, 15 automated 5' nuclease PCR, 24 and random amplified polymorphic DNA analysis, 43 which are major improvements in serovar determination, but the success of these assays requires highly technical skills, are prone to interlaboratory variation, are time consuming, and are incapable of processing large numbers of samples. Multiplex PCR assays are efficient platforms for detecting many gene targets in a single-sample preparation by incorporating multiple primer pairs. In the past, multiplex PCR assays were limited to identifying a small number of serovars, and these assays did not attempt to predict the overall pathogenicity of Salmonella isolates. 2,7,26,46 A recent study 27 described a multiplex PCR procedure consisting of 2 multiplex PCR panels for determining 30 clinically relevant serovars of Salmonella. The objective of the present study was to increase the number of serovars identified using the multiplex procedure and to develop an additional multiplex PCR panel called Salmonella Typing Virulence (STV) that determines the presence or absence of the genes spvC, invA, and sseL to allow researchers to predict the overall pathogenicity, invasiveness, and replication ability of the Salmonella isolates, respectively.
DNA microarrays, which work on nucleic acid hybridization principles, are widely used and have the multiplex capability to detect hundreds of thousands of genes in a single experiment.
8
Spotted DNA microarray platforms are cost effective, flexible, and easy to use in any laboratory with basic facilities and equipment.
11
This technology has been used in high-throughput detection of pathogens, such as methicil-lin-resistant Staphylococcus in hospitals,
44
and in the identification of antimicrobial resistance genes.
16,36
More recently, 3 studies
29,42,50
have focused on molecular serotyping of Salmonella using microarray protocols. These microarrays could identify only a limited number of serovars and required multiple probe sets for each serovar. For example, 414 probes and a software program were necessary to identify 14 different serovars.
42
Because there are presently 1,531 Salmonella enterica subspecies enterica group I serovars,
19
such assays can get complicated and expensive. Therefore, the objective of the current study was 1) to develop an economical, high-throughput, adaptable microarray that is capable of determining a large number of common serovars using a limited number of probes; and 2) to compare the results with a modified multiplex
Materials and methods Bacterial strains, culture, and DNA extraction
Salmonella isolates used in the present study were acquired from human clinical sources or were isolated from the feces of feedlot cattle in the midwestern United States or from various other sources, including turkey, swine, horse, and reptiles. Isolates were sent to Kansas State University Veterinary Diagnostic Laboratories (Man-hattan, KS) for species and serogroup identification and later to the National Veterinary Services Laboratories (NVSL) for serotyping by traditional antibody-based methods, using the Kauffmann-White scheme. Isolates were grown overnight at 37°C on tryptic soy agar, a and a single colony was picked and inoculated into 5 ml of tryptic soy broth a and grown at 37°C with shaking. Bacterial genomic DNA was extracted from 500 μl of overnight cultures using a commercial tissue kit b according to the manufacturer's instructions. Final DNA concentrations were determined using a spectrophotometer. c
STV multiplex PCR and primer design
A new multiplex panel for detecting virulence genes was developed. Primers to amplify the sseL gene 13,41 were designed using Primer 3 version 4.0 software, 4 based on conserved sequences from GenBank (accession AE008802), so that the melting temperature (Tm) and the product size were compatible with other reactions in the multiplex panel (Table. 1). Potential primer candidates were checked with BLAST searches for nucleotides until an acceptable set was found. The primers PT4 (amplifying sequences of a Salmonella Enteritidis phage type 4 strain) and STM 7 (amplifying sequences of a Salmonella Typhimurium strain) and sseL were combined with primers for spvC and invA, to create a unique multiplex PCR (STV). All primers included in this panel were analyzed using AutoDimer version 1 software e to ascertain that they did not form homodimers or heterodimers. 47
Multiplex PCR reaction protocols
The multiplex PCR was modified based on a previous study. 27 The protocol consisted of 2 multiplex PCR assays each consisting of 5 primer sets. The primer sets corresponded to arbitrary regions on the genome of S. Typhimurium 33 (STM) and Salmonella Typhi 35 (STY), as determined by microarray analyses. 27 The completed multiplex PCR reactions were electrophoresed on agarose gels, and the amplicons were numbered 1 through 5, based on their sizes (1 being the largest amplicon). If the PCR products were detected at a predicted location on a gel, it was considered positive for that reaction. For example, in the STM multiplex PCR reaction, if products corresponding to STM 1 and STM 5 primer sets amplified, then the amplicon code for the STM reaction for that particular isolate would be designated 1, 5. Similar amplicon codes were also generated for the STY multiplex panel. Occasionally, faint, nonspecific bands appeared (consistent with the previous study 27 ); however, only the most prominent bands were considered for amplicon coding. The incidences and the prominence of the faint, nonspecific bands were considerably reduced when the Qiagen Multiplex PCR kit b was used.
The STM assay was modified to use the Qiagen Multiplex PCR Kit b per the manufacturer's instructions. Briefly, 12.5 μl of 2X buffer was added to 4 μl of Q Solution with 50-100 ng of template DNA for a final volume of 25 μl. The reaction conditions included initial denaturation step at 94°C for 15 min, followed by 35 cycles of 94°C for 30 sec, 62°C for 45 sec, a step down to 58°C for 45 sec and 72°C for 1 min, and a final extension of 72°C for 5 min. All reactions were performed on a gradient thermocycler. f
The STY assay was modified to contain 1.25 U of Hot Start Taq, g 1.6X reaction buffer, 0.2 mM of each deoxyribonucleotide triphosphate (dNTP), and 50-100 ng of template DNA for a final volume of 50 μl. The STV reaction contained 1.25 U of Taq polymerase, h 1 × Ex Taq reaction buffer, g 0.2 mM of each dNTP, and 50-100 ng of template DNA for a final volume of 50 μl. Both the STY and STV used the same cycling parameters: 1 cycle of 94°C for 6 min; followed by 35 cycles of 94°C for 30 sec; a step up to 62°C for 15 sec, 58°C for 15 sec, and 72°C for 1 min; and a final extension of 72°C for 5 min.
Sample analysis and scoring of multiplex PCR products
The PCR products were separated by electrophoresis in a 3% agarose gel h in 1X TBE (90 mM Tris, 90 mM borate, 20 mM ethylenediamine tetra-acetic acid [pH 8.0]) for approximately 2 hr at 5.6 V/cm. The gel was stained using ethidium bromide (10 mg/ml), visualized under ultraviolet light, h and captured by a digital imaging system. i The PCR amplicon sizes were determined by comparison to molecular weight markers j and scored as described above.
List of primers used in the Salmonella Typing Virulence multiplex polymerase chain reaction assay.*
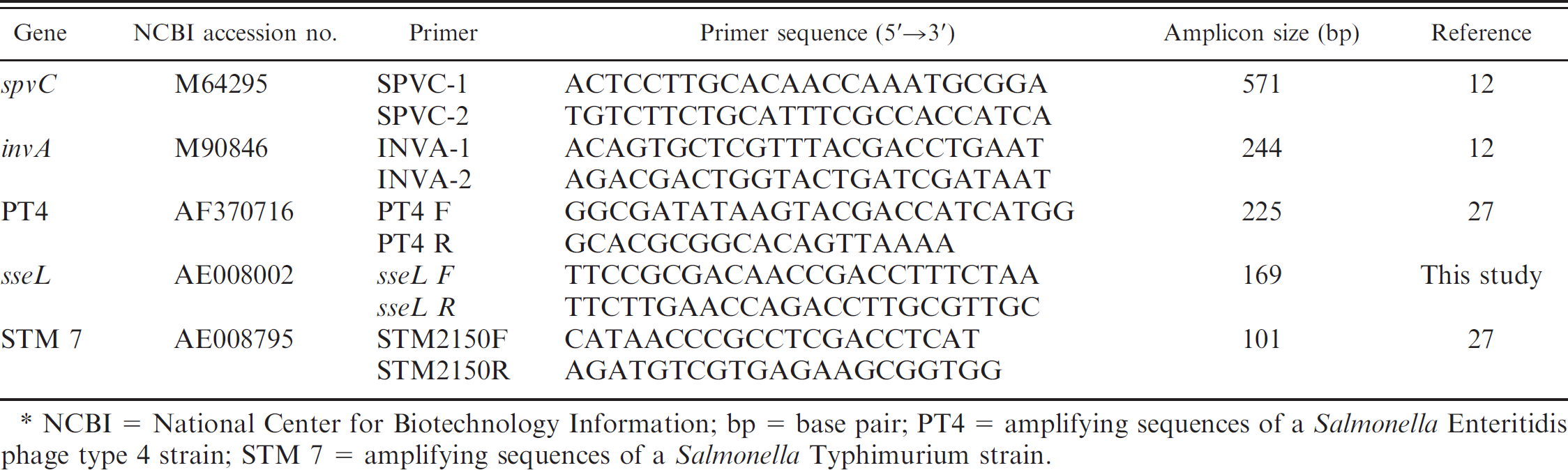
NCBI = National Center for Biotechnology Information; bp = base pair; PT4 = amplifying sequences of a Salmonella Enteritidis phage type 4 strain; STM 7 = amplifying sequences of a Salmonella Typhimurium strain.
Microarray probe design and printing
Initially, 3-8 candidate probes for each gene target were selected (a total of 63 probes) and printed in replicates of 3 or 10. These probes corresponded to genetic regions determined to be important for serovar differentiation by previous studies and with the multiplex PCR assays discussed above. 27,39 These genetic regions were originally chosen because heterogeneity in the region was reported to be helpful in differentiating between common serovars of Salmonella. 39 Based on their sensitivities, intensities of signals, and correlations with multiplex PCR data, 37 probes were selected (Table. 2) and were considered for further studies. The remaining 26 probes were excluded during the validation step (described later) from further consideration because of low intensity and/or negative hybridization, false positives and/or increased background, or signal inconsistency (data not shown). Candidate oligonucleotides were designed using OligoWiz 2.0 48 and synthesized. k On each microarray, the positive hybridization controls EUB (16S region conserved in all Eubacteria) 49 and rpoB 1 were included. Negative controls, including H2O, hybridization buffer only, and a 25-base pair (bp) DNA probe without homology to any listing in GenBank, were also printed. 38 Two fields containing the 70-mer oligonucleotides were printed on slides, l using a slide printer, m at a concentration of 35 μM each in replicates of 3 or 10. After printing, the DNA was cross-linked in a crosslinker n at 600 mJ and stored in the dark at room temperature until used.
Microarray DNA labeling and hybridization
Extracted genomic DNA was labeled directly using a BioPrime® Plus Array CGH Genomic (Direct) Labeling System o per manufacturer's protocol with slight modifications. After random primer hybridization, an additional 1.5 μl of 1 mM cyanine (Cy3) or 1 mM Cy5-2'-deoxycytidine-5′-triphosphate p was spiked into the labeling mixture to improve dye incorporation and amplification. For all DNA labeling reactions, the efficiency of labeling was determined using the microarray feature on a spectro-photometer, c which measures the fluorescent dye incorporation in the sample DNA. The labeled DNA was mixed with an equal volume of 2X hybridization solution 7 q and was used for hybridization. The microarray chips were prehybridized in blocking solution (0.1% bovine serum albumin, 5X saline-sodium citrate [SSC], and 1% sodium dodecyl sulfate [SDS]) at 42°C for 1 hr with shaking, and then spun at 2,200 × g to dry. The labeled DNA mixture was hybridized overnight at 42°C, followed by washing for 10 min in each of the following buffers: 10X SSC, 0.2% sodium lauroyl sarcosinate; 10X SSC; and 0.2X SSC. Lastly, the slides were quickly dipped in water, spun dry at 2,200 × g, and visualized on a slide reader. r
Other in vitro labeling kits, including ARES Alexa Fluor® 546 and 647 DNA Labeling Kits, o Array 900DNA™ Labeling Kit for DNA, q and DNA indirect labeling kits, o were employed, but the BioPrime system o was used in further experiments because it provided consistently higher efficiency of dye incorporation, as evidenced by a spectrophotometer c and by total spot fluorescence, on a microarray chip, as measured by a slide reader. r
Validation of molecular methods
For the multiplex blind study, 111 Salmonella culture samples and 31 bacterial DNA samples were submitted to the authors' laboratory. All samples were processed with STM, STY, and STV multiplex PCR and scored as described above. For the microarray blind study, 20 Salmonella pure culture samples, and 36 bacterial DNA samples were submitted to our laboratory. All samples were hybridized to serotyping microarray chips, and STM, STY, and STV scoring was conducted. Serotyping results were compared against results from traditional antibody-based serotyping, and statistical analyses (see below) were conducted.
Data analyses
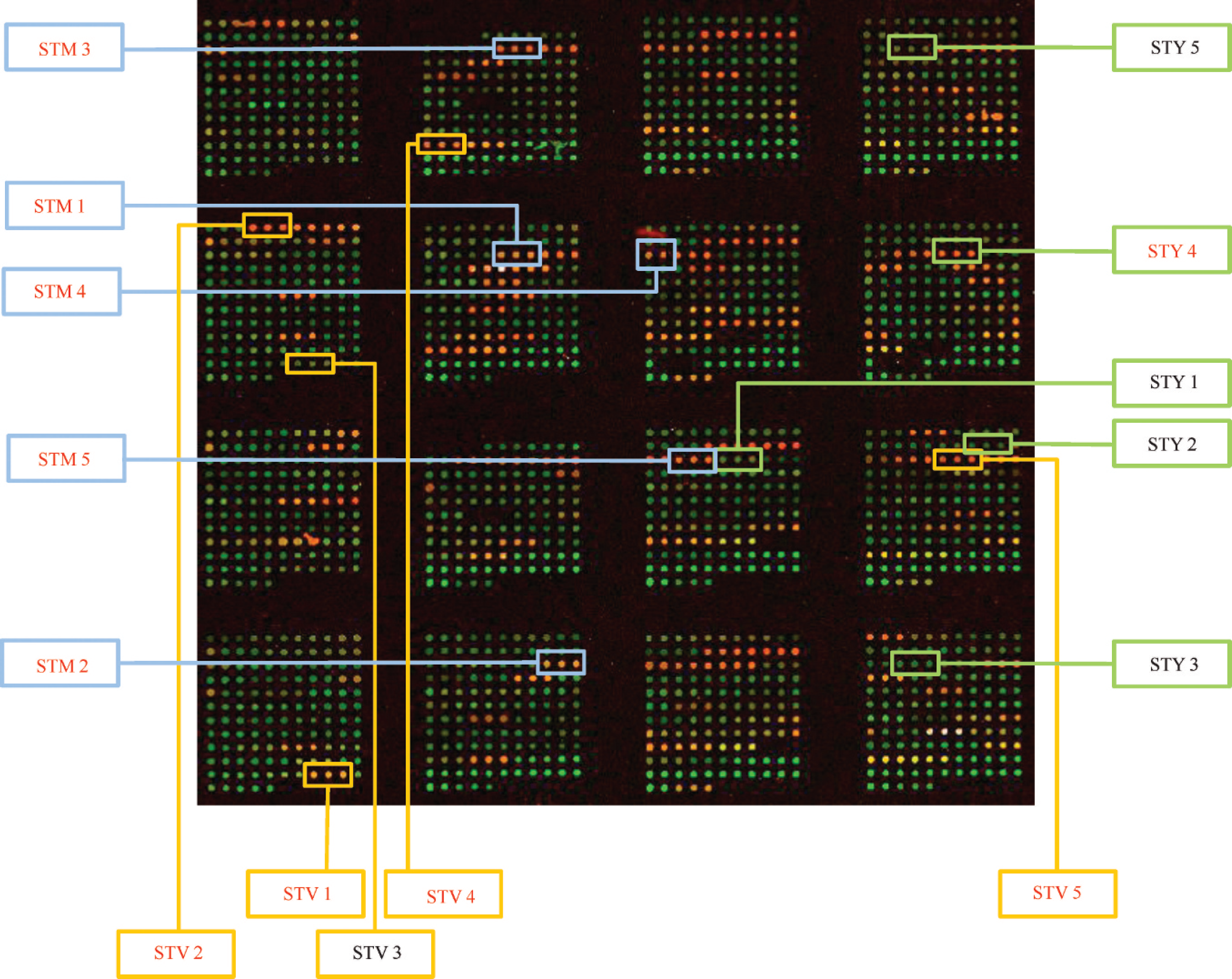
The fluorescent hybridization signals from the array were visualized using a slide reader r and matched to the GenePix Array List (GAL) file previously created by the microarray slide printer. A GenePix Report (GPR) file was generated measuring the overall intensities. Each target region was represented by 3 or 10 replicate spots. The fluorescence signals from each set were averaged and used for further analyses. Data were analyzed using the TIGR Multi-Experiment Viewer (TMEV) program s with 1 color setting. Data was also analyzed using a relative pathogen signal (RPS) ratio method previously described. 45 Briefly, the averaged signal intensities for each probe were divided by the signal intensities of the positive control spots to attain an RPS ratio. Mean hybridization signals to spots containing a unique DNA probe (25 bp) that shared no significant homology to sequences in GenBank were subtracted from the RPSs of test spots. 38 A final ratio was determined from that: Values greater than 0.25 were considered positive, and values less than 0.25 were considered negative.
The 70-mer probes used in the current study for serotyping Salmonella enterica. *

NCBI = National Center for Biotechnology Information; STM = Salmonella Typhimurium; STY = Salmonella Typhi; STV = the Salmonella Typing Virulence multiplex polymerase chain reaction panel.
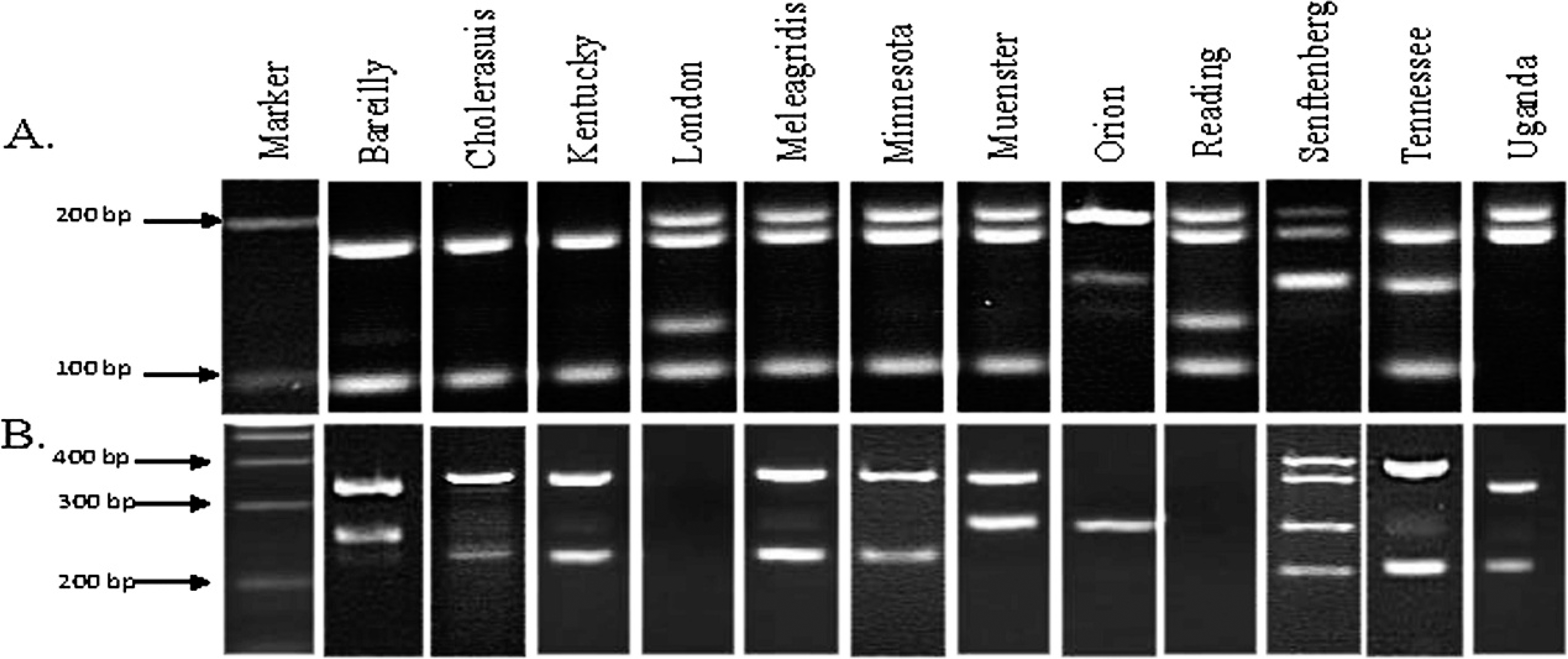
Salmonella Typhimurium (STM; A) and Salmonella Typhi (STY; B) multiplex polymerase chain reaction (PCR) products of 12 previously unscreened serovars. If PCR products (5 primer sets for STM and 5 primer sets for STY) were present at the predicted location on a gel, it was considered positive for those reactions. An amplicon code was designated based on the bands that were present. For example, serovar Bareilly (panel A, second lane) had PCR products for primer sets STM 2 and STM 5. Therefore, the amplicon code for STM multiplex PCR for Bareilly is 2, 5 (see Table 3 for amplicon codes for all serovar tested in the current study).
The sensitivity and specificity of the multiplex and the microarray were calculated using the antibody-based testing as the gold standard. Specificity was measured as the number of true negatives/(number of true negatives + false positives), and sensitivity was measured as the true positives/(number of true positives + false negatives).
Results Multiplex PCR assay development and validation
The newly developed multiplex PCR panels successfully identified 42 serovars of Salmonella. This included Bareilly, Choleraesuis, Kentucky, London, Meleagridis, Minnesota, Muenster, Orion, Reading, Senftenberg, Tennessee, and Uganda (Fig. 1; Table 3), as well as the 30 serovars published previously 27 (data not shown).
A third multiplex STV panel containing 5 sets of PCR primers was created to predict the virulence of a Salmonella isolate and to further discriminate some serovars (Fig. 2). The amplicons for sseL (169 bp) and invA (244 bp) were present in all isolates tested. As previously noted, invA has been used as a genetic marker for all pathogenic Salmonella. 12 The PCR targeting the spvC gene amplified a 571-bp product from serovars Choleraesuis, Dublin, Enteritidis, and Typhimurium only (Fig. 2). Amplicon codes were generated as described above, for all serovars tested, including the 12 new serovars (Tables 1, 2).
In the blind study, the multiplex PCR-based serovar determination correlated with the traditional antibody-based serotyping results (performed by NVSL) 100% of the time (total of 111 isolates) as belonging to 23 serovars when their serogroup was known. In the absence of the serogroup data, the multiplex PCR assay successfully identified the serovars of 135 out of 142 Salmonella isolates, in which case the overall sensitivity and specificity were both 95.3%.
Microarray serovar identification
A spotted microarray platform was developed containing 70-mer probes for STM, STY, and STV genes. Based on the hybridization patterns, unique signatures were developed for the tested serovars. This array of 37 probes was successful in determining all serovars of Salmonella tested (total of 86 isolates belonging to 40 serovars; 10 of these isolates are shown in Fig. 3). The correlation between the signals generated in the microarray and the bands amplified in multiplex PCR matched 30 out of the 30 isolates tested in the initial validation step (data not shown).
Complete list of all amplicon codes for Salmonella Typhimurium (STM), Salmonella Typhi (STY), and the Salmonella Typing Virulence multiplex polymerase chain reaction panel (STV).*
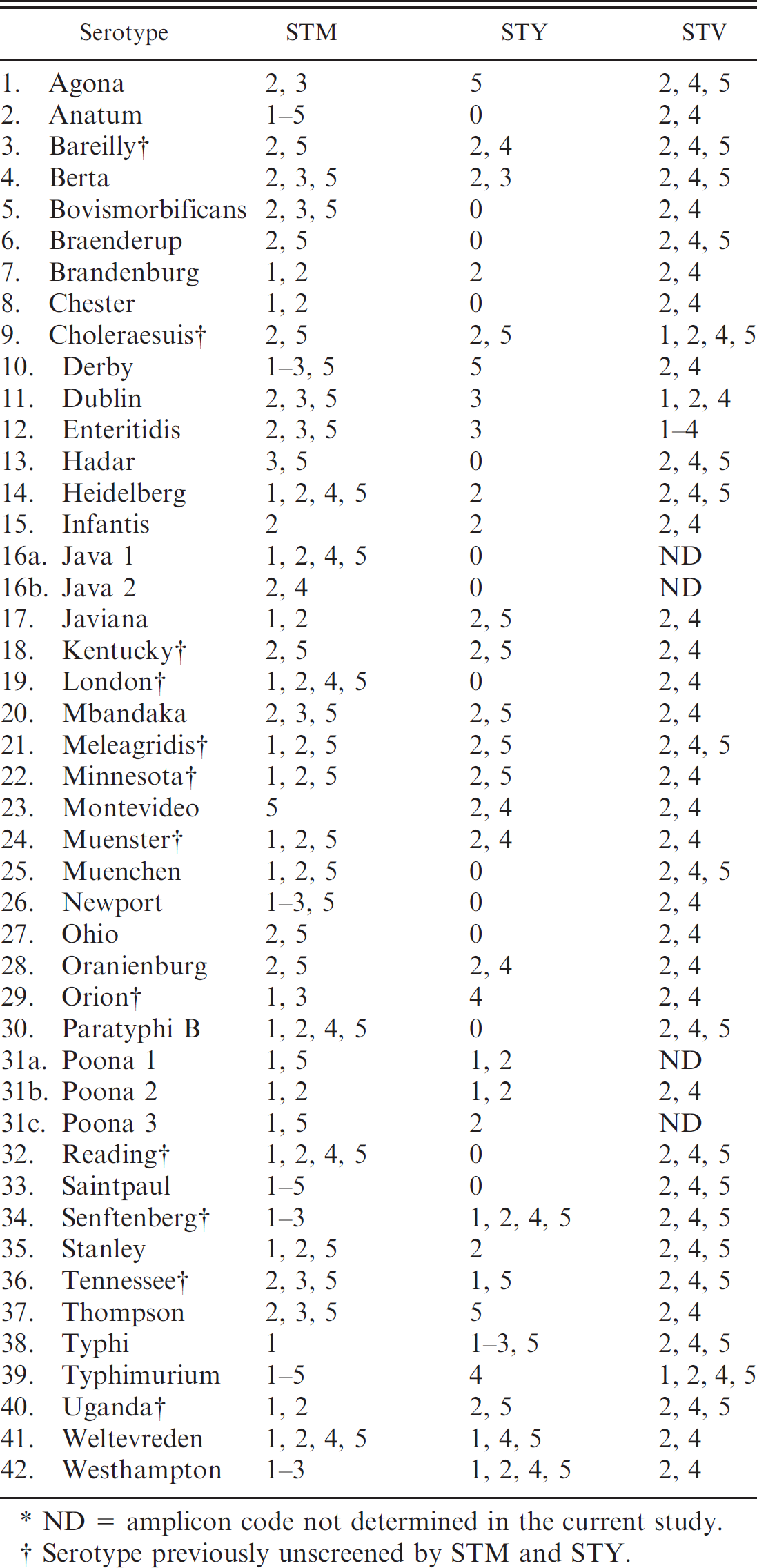
ND = amplicon code not determined in the current study.
Serotype previously unscreened by STM and STY.
In the blind study, serovars of 52 out of 56 total isolates representing 28 different serovars (Table. 4) were successfully identified. The sensitivity and specificity of the microarray-based serotyping assay were determined to be 93.3%. Because many of the isolates used in the blind study were submitted as DNA samples from diagnostic laboratories throughout across the country, the serogroup data were unknown.
Discussion
In the current study, the discriminatory power of a multiplex PCR assay for molecular serovar identification of Salmonella was greatly improved from the existing techniques 27 with the addition of a new multiplex PCR panel (STV) that not only incorporates 2 gene targets (STM 7 and PT4) that differentiate certain serovars 27 but also detects the presence of the virulence markers spvC, invA, and sseL. All isolates tested were positive for virulence genes invA and sseL. Because only strains of Salmonella isolated from clinical sources were included in the present study, the presence of these genes in less-virulent strains, especially those from environmental sources, is not known.
The invA gene encodes an invasion protein and has been reported to be present in most strains of Salmonella isolated from animals and humans. 31 The sseL encodes a deubiquitinase enzyme that contributes to intracellular survival and invasion by Salmonella in macrophages and is claimed to be present only in highly virulent strains. 13,41 The virulence gene spvC is generally carried in a plasmid but may also be present in the genome and has been shown in previous studies to improve survival in the presence of starvation stress in the host tissues. 6,12,20,28 The spvC gene encodes a phosphothreonine lyase that has significant similarity to OspF of Shigella flexneri and has an inhibitory effect on signal transduction mediated by ERK and JNK pathways and NFkB activation in eukaryotic cells, thereby, inhibiting proinflammatory cytokine responses. 32 Previous studies have reported that the spvC gene is predominant in 4 serovars tested, including Typhimurium, Cholerae-suis, Dublin, and Enteritidis, 12,20 with Salmonella Typhimurium and Enteritidis being the most common serovars isolated from diarrheal patients. 20 Therefore, the presence of the spvC gene may predict the ability of a Salmonella strain to survive in a host environment, its capacity to infect other hosts, and the possibility of causing an outbreak. The potential of the assay was demonstrated by testing of 111 blinded isolates with a sensitivity of 100% when serogroup data were available. When calculations were performed without considering the serogroup data (135 of 142 isolates), the sensitivity and specificity was determined to be 95.3%.
Microarrays have been used previously to identify serovars of bacteria such as Escherichia coli, 3,21 Salmonella, 9,18,29,30,39,50 and Bartonella 5 because of the rapidity of the assay and its ability to provide high throughput and consistently reproducible results. In addition, the charged coupled device detection of fluorescent spots in microarrays has been reported to provide high sensitivity. 17 The microarray developed in the current study consisted of probes that targeted the genetic regions shown to be important for serovar differentiation by previous studies 39 and multiplex PCR assays. The data from the microarray-based serotyping were compared with analysis of the same isolates using a multiplex PCR, and the sensitivity and specificity was determined to be 93.3% (see Materials and Methods section). A previous multiplex PCR study 2 noted that some target DNA may not be amplified because of intraserovar variation at the primer binding regions, which could lead to the compiling of false-amplicon codes. In the present study, the 95.3% concordance of multiplex PCR and microarray suggests that such mutations may not significantly affect the effectiveness of multiplex PCR to determine the serovar of a Salmonella isolate.
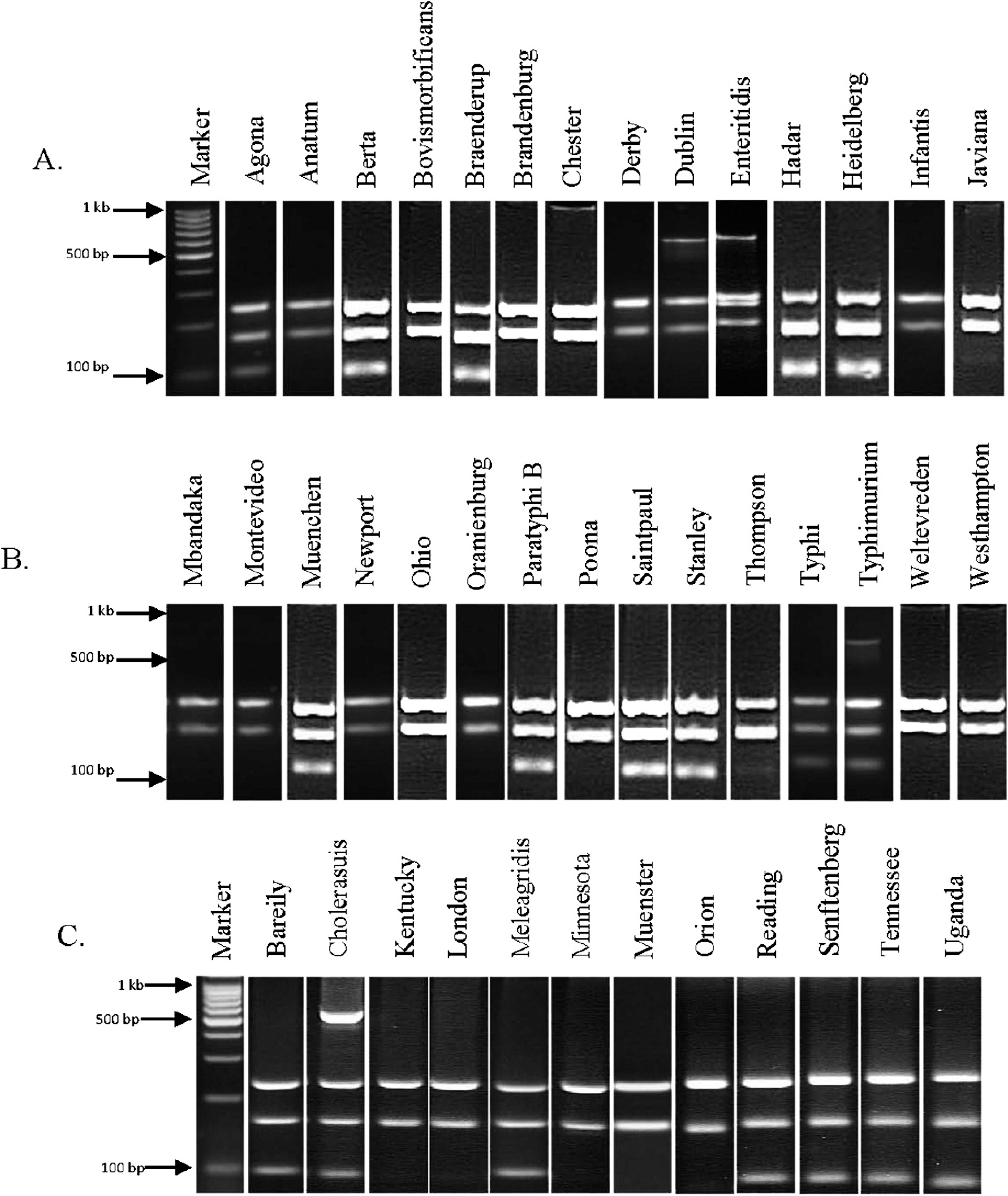
Multiplex polymerase chain reaction (PCR) Salmonella Typing Virulence (STV) products of 29 serovars for which A, Salmonella Typhimurium (STM) and B, Salmonella Typhi (STY) were screened previously. 27 C, multiplex PCR STV products of the 12 new Salmonella enterica serovars. An amplicon code was designated based on the bands that were present. For example, serovar Agona (panel A, second lane) had PCR products for primer sets STV 2, STV 4, and STV 5. Therefore, the amplicon code for STV multiplex PCR for Agona is 2, 4, 5 (see Table 3 for amplicon codes for all serovars tested in the current study).
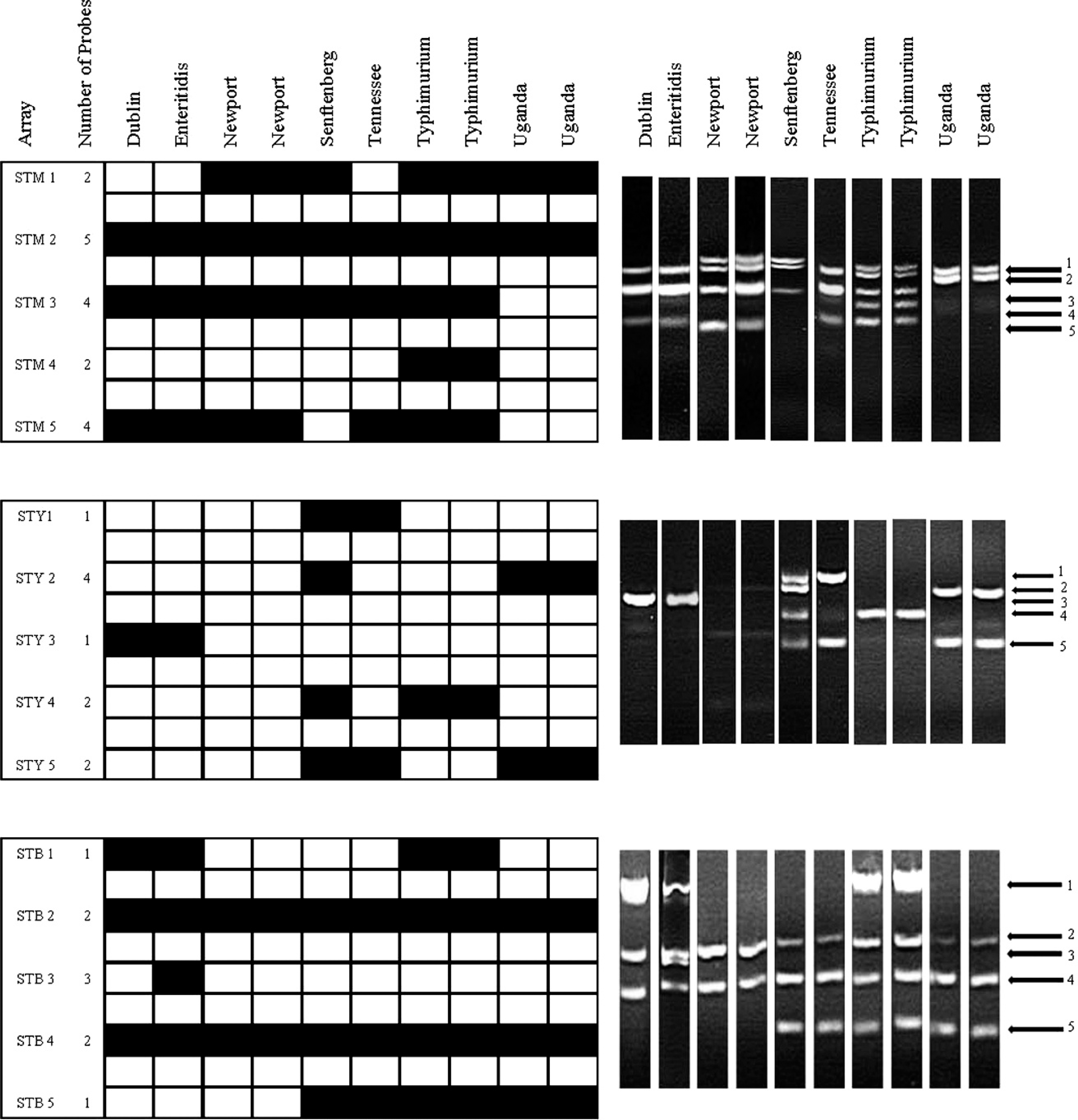
Results from microarray (black is positive, and white is negative) and multiplex polymerase chain reaction for Salmonella Typhimurium (STM) 1-5, Salmonella Typhi (STY) 1-5, and Salmonella Typing Virulence (STV) 1-5 genes of 10 Salmonella isolates representing 7 serovars.
The current study describes 2 rapid molecular methods that can accurately identify the serovars of common clinical isolates of S. enterica subsp. enterica. The multiplex PCR is straightforward and can currently be applied in any laboratory with access to PCR and gel electrophoresis equipment. The growing number of Salmonella genome sequences available for analysis and comparison by complete genomic hybridization and other methods will identify future targets to improve the discrimination of serovars that may share the same amplicon codes. Of the 1,531 serovars of Salmonella enterica currently known, 19 the blind studies in the present study only covered a relatively limited number of them. Although previous studies and those conducted earlier 27 included isolates from some of the most common serovars encountered, 20 larger validation studies will be necessary to use these tests as diagnostic tools. In addition, higher throughput for diagnostic samples may be achieved by performing a fluorogenic 5' nuclease PCR assay.
Complete list of all serotypes tested in the current study with multiplex polymerase chain reaction (PCR) and microarray in the blind study.*
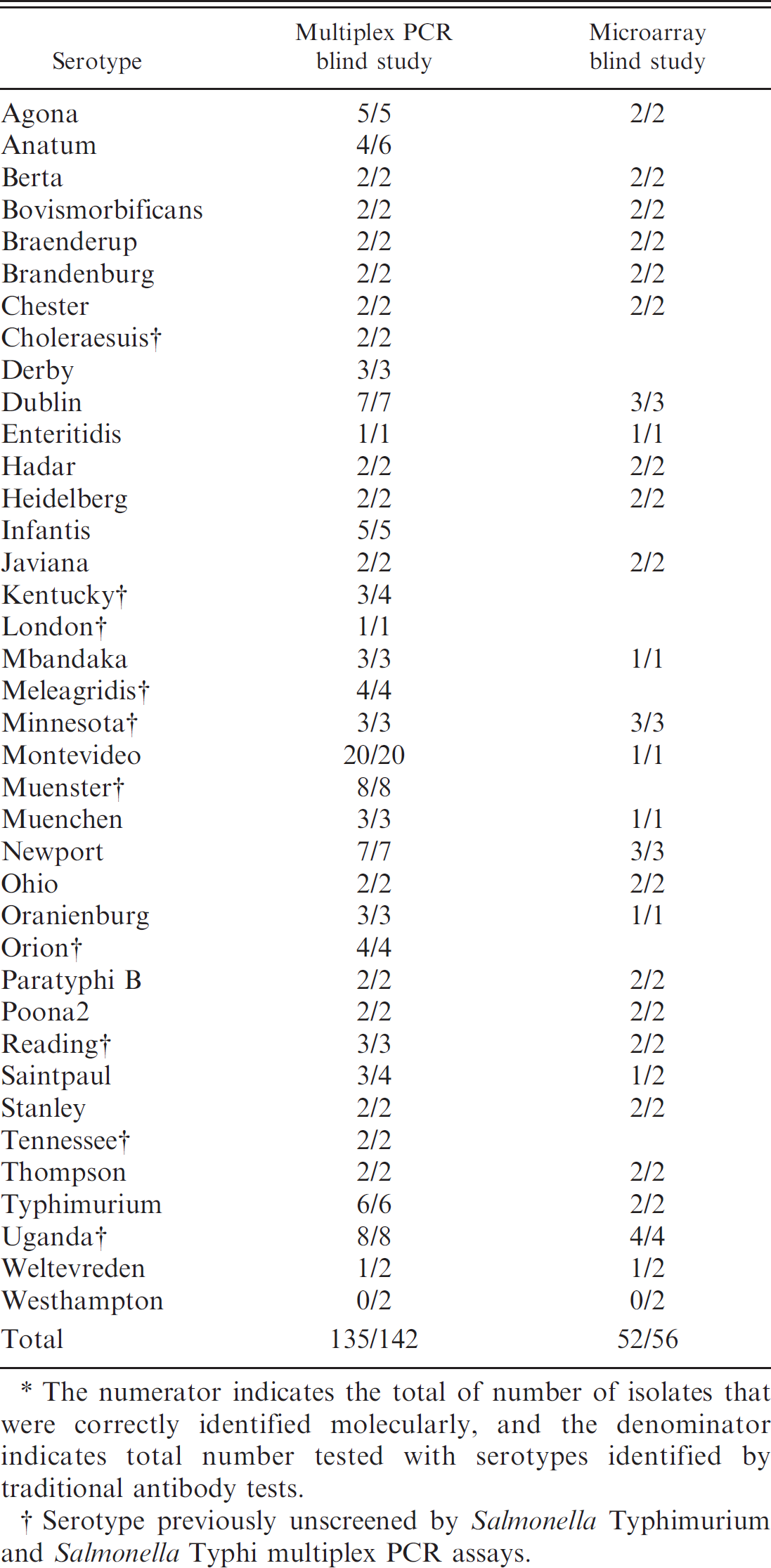
The numerator indicates the total of number of isolates that were correctly identified molecularly, and the denominator indicates total number tested with serotypes identified by traditional antibody tests.
Serotype previously unscreened by Salmonella Typhimurium and Salmonella Typhi multiplex PCR assays.
All blind samples (n = 142) were processed for multiplex PCR, and a subset (n = 56) were also tested by microarray analysis (Table. 4). The smaller number of samples used in the microarray blind studies were due to the relatively higher costs to perform the assay. Conventional antibody-based serotyping has become increasingly expensive in the recent past because of high costs associated with licensing and transportation of live or frozen cultures via mail, maintenance of freezers for storing the cultures, and production of large collections of specific antisera. Expense of the microarray can be reduced by low-density arrays with support matrices, such as nitrocellulose; by multiple arrays printed on each glass slide and each array hybridized with DNA labeled by different fluorescent markers from different isolates; and by labeling targets with colorimetric, instead of fluorescent, dyes.
Although multiplex PCR arrays provide a molecular method for identifying the serovar of a Salmonella isolate, microarrays have the potential to acquire more than 1 million data points in a single experiment. Also, a microarray probe set can be easily incorporated into any established diagnostic micro-array protocol, thereby increasing the overall strength of that platform. For example, the authors have incorporated the serovar-identifying array in an antimicrobial-resistance gene array to provide a platform for evaluating resistance profiles of Salmonella serovars (see Supplemental Data). 37
The authors of the current study have identified multiplex PCR amplicon codes for 42 serovars and have determined microarray hybridization profiles for 40 serovars of Salmonella enterica. In the present study, a newly developed multiplex PCR reaction (STV), which detects virulence genes (sseL, invA, and spvC), is reported. The presence or absence of spvC was successful in identifying a subset of serovars that has been shown previously to cause significant human and animal diseases. It is believed that the tests developed in the present study will aid in understanding the epidemiology and diversity of Salmonella.
Acknowledgements
The authors would like to acknowledge the following people for their help during this work: David Renter, Megan Jacob, Jahangir M. Alam, and Tanya Purvis (Kansas State University) for help with isolate acquisition. The authors would also like to thank Amit Kumar (Kansas State University) for his help in editing of this manuscript. Microarray chip printing was performed in the Gene Expression Facility at Kansas State University. The facility is supported through the National Science Foundation grant DBI-0421427. Additional microarray printing was performed at the Microarray Core Facility, Department of Genetics Cell Biology and Anatomy, University of Nebraska Medical Center (UNMC), Omaha, NE. The UNMC Microarray Core Facility receives partial support from National Institutes of Health grant P20 RR016469 from the IDeA Networks of Biomedical Research Excellence (INBRE) Program of the National Center for Research Resources. This study is published as contribution 08-000-J from the Kansas Agricultural Experiment Station. The mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
a.
Difco, Becton Dickinson, Sparks, MD.
b.
DNeasy® Tissue Kit, Qiagen Inc., Valencia, CA.
c.
NanoDrop® ND-1000 spectrophotometer, Thermo Fisher Scientific Inc. (NanoDrop products), Wilmington, DE.
f.
Mastercycler® gradient thermocycler, Eppendorf North America, Hauppauge, NY.
g.
Takara Bio Inc., Otsu, Shiga, Japan.
h.
Fisher Scientific Co., Pittsburgh, PA.
i.
Fotodyne Inc., Hartland, WI.
j.
100-bp ladder; Promega Corp., Madison, WI.
k.
Operon Biotechnologies Inc., Huntsville, AL.
l.
Ultra Gap, Corning Inc., Corning, NY.
m.
QArray2 System, Genetix Ltd., New Milton, Hampshire, United Kingdom.
n.
Stratalinker® UV Crosslinker 2400, Stratagene, La Jolla, CA.
o.
BioPrime® Plus Array CGH Genomic Labeling System, Invitrogen Corp., Carlsbad, CA.
p.
Amersham Biosciences, Piscataway, NJ.
q.
Genisphere Inc., Hatfield, PA.
r.
GenePix 4000B slide reader, Molecular Devices, Sunnyvale, CA.
s.
The Institute for Genomic Research, Rockville, MD.