
Abstract
A newly developed polymerase chain reaction (PCR)-based method to rapidly and specifically detect Geomyces destructans on the wings of infected bats from small quantities (1–2 mg) of tissue is described in the current study (methods for culturing and isolating G. destructans from bat skin are also described). The lower limits of detection for PCR were 5 fg of purified fungal DNA or 100 conidia per 2 mg of wing tissue. By using histology as the standard, the PCR had a diagnostic specificity of 100% and a diagnostic sensitivity of 96%, whereas the diagnostic sensitivity of culture techniques was only 54%. The accuracy and fast turnaround time of PCR provides field biologists with valuable information on infection status more rapidly than traditional methods, and the small amount of tissue required for the test would allow diagnosis of white-nose syndrome in live animals.
Introduction
Emerging wildlife diseases constitute a new and potentially devastating threat to animal populations worldwide. 4,12,13 In 2007, biologists discovered a new disease that affected hibernating bats of east-central New York. 2 By winter 2009, it had spread as far as southern Virginia, with population declines in affected caves approaching 100%. 20 The disease, characterized by fungal growth on the wings, tail, ears, and muzzles of bats, was named white-nose syndrome (WNS). The fungus that causes the hallmark cutaneous infection of WNS has been described as the new species Geomyces destructans, 7 and the disease is currently diagnosed by confirming skin infection by the fungus. 14
At present, G. destructans infection is verified through fungal culture and/or histologic examination of bat wing, ear, and/or muzzle tissue(s). Fungal culture is complicated by the nonsterile nature of bat skin, which makes axenic isolation difficult. Histology is more sensitive but is labor intensive and requires specialized training for interpretation. In addition, both methods have turnaround times of more than 1 week and often require larger amounts of tissue than can reasonably be collected from a live bat.
Because WNS continues to spread across eastern North America, there is an urgent need for a simple and rapid test to confirm infection. Polymerase chain reaction (PCR) offers a fast, reliable, and economical alternative to histology and culture, and is quickly becoming the most widely used method for detecting human, animal, and plant pathogens. 11,15,19,21,23 The current study describes a newly developed PCR-based test to detect the WNS-associated fungus G. destructans directly from bat wing tissue within hours. Reliability of the PCR method was assessed by comparisons with culture and histology techniques.
Materials and methods
Samples
Bat carcasses were submitted to the U.S. Geological Survey–National Wildlife Health Center (Madison, WI) for diagnostic evaluation in 2008 and 2009. Animals either were found dead or were euthanized before submission. For the present study, 78 carcasses were analyzed, including submissions from 35 collection events in 13 states (Alabama, Connecticut, Indiana, Maryland, New Hampshire, New Jersey, New York, Ohio, Pennsylvania, Virginia, Vermont, Wisconsin, and West Virginia). Eight species of bats were represented, including the big brown bat (Eptesicus fuscus), gray bat (Myotis grisescens), eastern small-footed bat (Myotis leibii), little brown bat (Myotis lucifugus), northern long-eared bat (Myotis septentrionalis), Indiana bat (Myotis sodalis), evening bat (Nycticeius humeralis), and tricolored bat (Perimyotis subflavus).
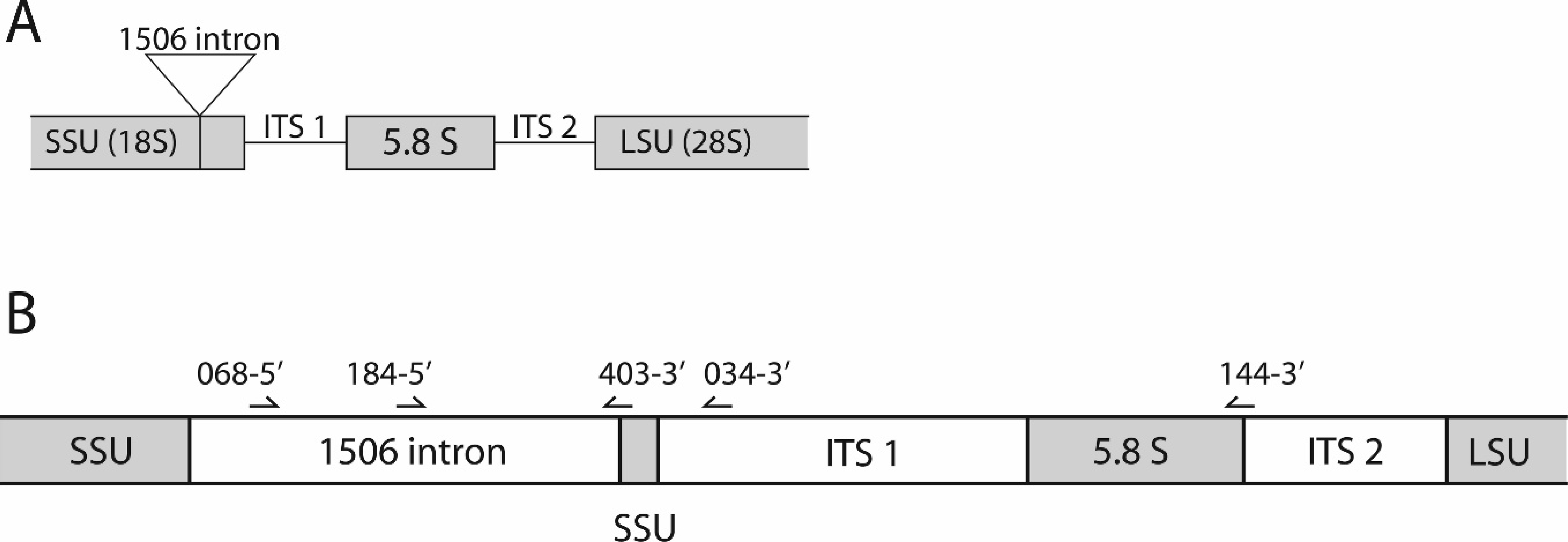
Primer pairs developed for amplifying fragments of the Geomyces destructans ribosomal RNA gene internal transcribed spacer (ITS) region. *

Tm = melting temperature; nu = nuclear; SSI = small subunit intron; Gd = Geomyces destructans; nt = nucleotide; SSU = small subunit.
The Tm values were calculated by using sequence analysis software.
The primer pair that consisted of nu-SSI(1506)-184-5'-Gd and nu-5.8S-144-3'-Gd was used for the analyses described herein.
Histology
To determine whether bats met the diagnostic criteria for WNS, rectangular pieces of wing membrane, approximately 1.5 cm × 3.0 cm, were cut and prepared as described previously. 14 Cross sections of the rolled wing skin then were examined microscopically for the presence of distinctive G. destructans conidia in conjunction with fungal hyphae, cup-like epidermal erosions, ulcers, and invasion of underlying connective tissue.
Geomyces destructans culture technique
To isolate G. destructans from bat skin, a piece of wing tissue approximately 1.5 cm × 1.5 cm was removed from each bat and placed flat onto Sabouraud dextrose medium that contained chloramphenicol and gentamycin. a Plates were incubated at 7°C for 10–30 days and were examined every 1–3 days. Fungal growth that resembled G. destructans was isolated and identified by observation of the distinctively curved single-celled conidia 7 by using a 40x objective. The internal transcribed spacer (ITS) region of the ribosomal RNA (rRNA) gene of select isolates was sequenced to confirm identification; all sequenced isolates were identical to G. destructans type isolate 20631-21 7 (GenBank accession no. FJ231098).
For PCR controls, pure cultures of the G. destructans type isolate were grown at 7°C on Sabouraud dextrose medium that contained chloramphenicol and gentamycin. Cultures were maintained by transferring mycelial plugs to fresh growth medium every 60–90 days.
DNA extraction
For genomic DNA (gDNA) isolation, a piece of wing membrane that measured approximately 3 mm × 3 mm (1–2 mg) was excised adjacent to the wing section used for culture. Larger samples inhibited PCR, presumably because of excessive amounts of nucleic acid or other inhibitors present in the wing tissue. DNA was extracted by using a commercial gDNA purification kit b per the manufacturer's instructions (solid tissues protocol), with the following amendments: proteinase K was added to a final concentration of 0.5 mg/ml during the cell lysis procedure and no RNase treatment was performed. Final DNA concentration was measured by using a spectro-photometer c and was diluted, if necessary, to a final concentration of 10–40 ng/μl. Genomic DNA was extracted from pure cultures of G. destructans, d and the concentration was measured by using a commercial DNA assay kit. e
Primer design, PCR, and sequencing
All small subunit (SSU; 18S) rRNA gene sequences from G. destructans examined to date (n = 28) contained a 414-nt (nucleotide) intron (GenBank accession no. EU884924 plus unpublished sequence data) at position 1506, as previously defined. 6 This relatively variable sequence area located within the conserved SSU rRNA gene was used for PCR primer design. Two forward primers, nu-SSI(1506)-068-5'-Gd and nu-SSI(1506)-184-5'-Gd, are located within the 1506 intron. Three reverse primers were also designed. One, nu-SSI(1506)-403-3'-Gd, bridges the 5' end of the 1506 intron and the adjacent SSU rRNA gene; the second, nu-ITS1-034-3'-Gd, is within the ITS1 region; and the third, nu-5.8S-144-3'-Gd, bridges the 3' end of the 5.8S gene into the adjacent ITS2 region (Table 1, Fig. 1). Primer names follow the nomenclature previously established 5 by using nu for nuclear, SSI for small subunit intron, the number 1506 to indicate the intron insertion position based upon the sequence of designated type isolate 20631-21 (GenBank accession no. FJ231098), and Gd to designate G. destructans. All 6 possible primer pairings were tested, and the pair that consisted of nu-SSI(1506)-184-5'-Gd and nu-5.8S-144-3'-Gd was chosen for diagnostic applications because it yielded the strongest and most-consistent PCR product.
Polymerase chain reaction was conducted by using DNA polymerase f per the manufacturer's instructions. Reactions included 1 μl of extracted DNA (10–40 ng/μl) in a volume of 50 μl. Cycling conditions consisted of an initial denaturation at 98°C for 2 min, followed by 40 cycles of 98°C for 10 sec, 50°C for 30 sec, and 72°C for 1 min, and a final extension at 72°C for 7 min. Five microliters of PCR product was loaded onto a 2% agarose gel that contained a nucleic acid gel stain g and was run at 110 V for 20 min by using a 50 base pair (bp) or 100-bp PCR ladder. h Generation of a 624-nt fragment confirmed the presence of G. destructans DNA in the sample.
Amplification products from samples identified as PCR positive for G. destructans were sequenced to confirm specificity of the method. When multiple bats from a single location were positive by PCR, amplification product from 1 randomly selected sample was sequenced. The PCR products were submitted to the University of Wisconsin–Madison Biotechnology Center DNA Sequencing Facility (Madison, WI) for direct, double-stranded sequence determination by using a commercial DNA sequencing system. i Reaction products were analyzed by using an automated DNA sequencer. i Complementary strand sequencing reaction results were assembled and edited for accuracy by using sequence analysis software. j
Evaluation of the PCR assay
To determine the DNA detection limit of the PCR, 50-μl reactions were run by using purified G. destructans gDNA template in quantities that ranged from 0.05 fg to 50 ng. To determine the detection limit of the PCR for G. destructans conidia on bat wing skin, conidia from 3-month-old pure cultures of the fungus on Sabouraud dextrose medium were harvested by flooding with 10 ml of 1x phosphate buffered saline (PBS) solution that contained 0.5% Tween20 h (PBST). After gently swirling the plate for 30 sec, the liquid was collected and centrifuged at 6,000 × g for 5 min, and the pellet was washed once with PBST. Washed conidia were resuspended in PBST, enumerated by using a hemocytometer, and suspended in 50 μl PBST to final quantities of 100, 101, 102, 103, 104, and 105. Pieces of wing skin (3 mm × 3 mm; 2 mg) from a bat collected outside of the WNS-affected region and shown to be free of fungal infection by histology were then added to the conidial suspensions. DNA was extracted from each suspension for subsequent PCR analysis.
The diagnostic sensitivity and specificity of culture and PCR were compared by using histology as the standard. Diagnostic sensitivity was defined as the percentage of bats that were positive by PCR compared with histology and was calculated by using the formula Bp/(Bp + Bfn), in which Bp was the number of bats positive for G. destructans by PCR that were also histologically positive for WNS, and Bfn was the number of false-negative results (i.e., bats that were negative for G. destructans by PCR but positive by histology). Diagnostic specificity was defined as the percentage of bats that were negative by PCR that were truly free of WNS as verified by histology according to the formula Bn/(Bn + Bfp), in which Bn was the number of bats negative for G. destructans by PCR that were also negative for WNS by histology, and Bfp was the number of false-positive results (i.e., bats positive for G. destructans by PCR that were negative by histology).
Results
Histology is considered the standard for diagnosis of WNS and was used as the benchmark with which PCR and culture-based detection were compared. Forty-eight of the 78 carcasses examined for the current study (62%) had lesions on the wings consistent with the diagnostic criteria for WNS. 14
Culture frequently revealed the presence of multiple species of fungi and bacteria on the surface of the bat wings. By using culture technique, G. destructans was isolated from only 26 of the 78 animals tested (33%). All the bats from which G. destructans was cultured were also histologically positive for WNS. Thus, no false-positive results were obtained, and the diagnostic specificity was 100%. Compared with histology, the diagnostic sensitivity of culture was 54%.
Polymerase chain reaction detected the presence of G. destructans DNA in 46 of the 78 bats tested (59%). All the animals that were PCR positive for the fungus were also positive for WNS by histology, which resulted in a diagnostic specificity of 100%. With only 2 false-negative results, the diagnostic sensitivity of the PCR-based detection method was 96%. A comparison of the detection methods is presented in Table 2.
Comparison of the sensitivity and specificity of polymerase chain reaction (PCR)-based and culture-based methods to histopathology, defined as the standard for detecting the white-nose syndrome (WNS)-associated fungus, Geomyces destructans, on bat wings. *

The sample set (n = 78) consisted of 30 WNS-negative and 48 WNS-positive bats.
After PCR amplification, pure gDNA from G. destructans and extracted wing tissue samples identified as PCR positive yielded an intense band of 624 nt (Fig. 2); 2 additional faint bands of approximately 300 nt were also often observed. The 624-nt amplification products from 26 bats that represented all the PCR-positive collection sites were sequenced and shown to be 100% identical to the G. destructans rRNA gene region targeted for amplification. In 2 samples identified as PCR negative for G. destructans, PCR yielded a series of bands of equal intensity that were distinct from the predominant 624-nt band indicative of G. destructans DNA (data not shown). Histology confirmed that these bats did not have WNS.
When purified G. destructans gDNA was used as template, the limit of PCR detection was 5 fg (Fig. 3A); bands representative of the targeted PCR product decreased in intensity as the quantity of DNA decreased. When G. destructans conidia were cosuspended with bat wing skin before DNA extraction, the limit for PCR detection was 100 conidia per 2 mg wing tissue (Fig. 3B). Decreasing PCR product band intensity was again observed as the number of conidia decreased.
Discussion
The diagnostic specificity of the G. destructans PCR detection method was 100%. This is consistent with many other PCR-based detection techniques for fungal infections, 3,10,16,17 although lower values of 65–89% also were reported. 1,8,9,18,22 The diagnostic sensitivity of the PCR method was 96%. This value falls within the range of sensitivities (70–100%) reported for similar assays. 1,8–10,16–18,22 Diagnostic sensitivity of PCR (96%) was significantly greater than that of the culture method (54%). In only one instance did culture detect G. destructans when PCR did not. The low diagnostic sensitivity of culture is likely because of competition by other microbes present on bat skin that quickly out compete G. destructans when grown on solid medium, thereby masking its presence. Detection by culture also requires that viable fungus be present on the wing once it reaches the laboratory; histology and PCR both can detect the fungus in a nonviable state.
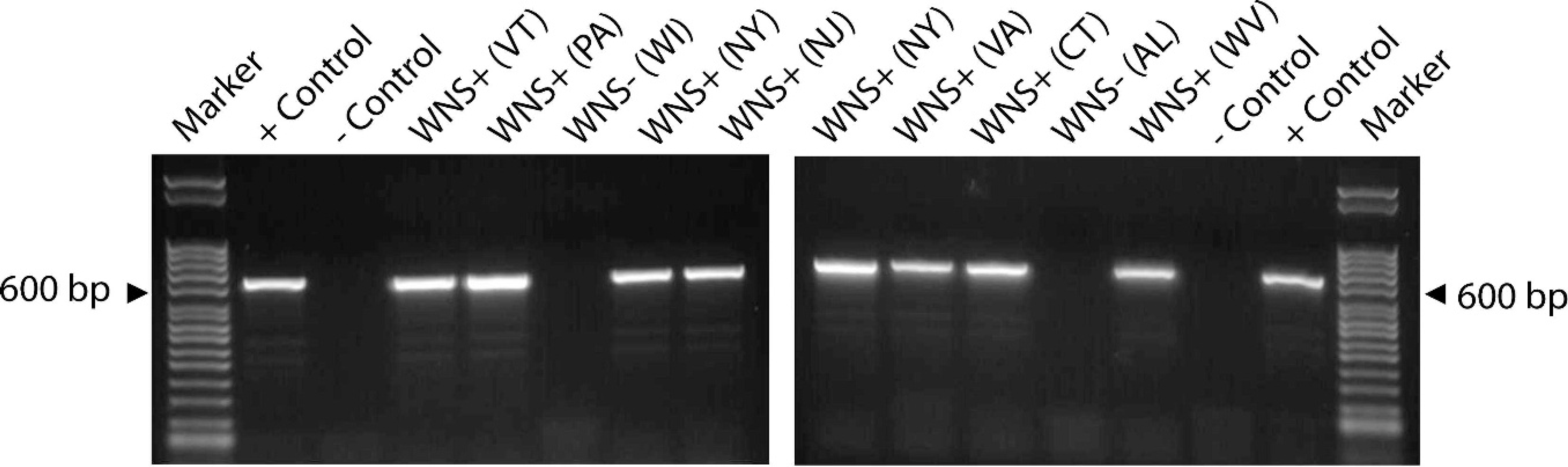
Polymerase chain reaction (PCR)-based detection of Geomyces destructans on bat wing skin by using primers nu-SSI(1506)-184-5'-Gd and nu-5.8S-144-3'-Gd. A 624-nucleotide band indicates the presence of G. destructans. Positive control reactions used genomic DNA extracted from pure cultures of G. destructans as template; negative controls had no template added. Histology results for each PCR reaction are indicated above the lanes as either white-nose syndrome (WNS)+ or WNS-; the state from which each animal was collected is identified in parentheses. nu = nuclear; SSI = small subunit intron; Gd = Geomyces destructans; bp = base pair.
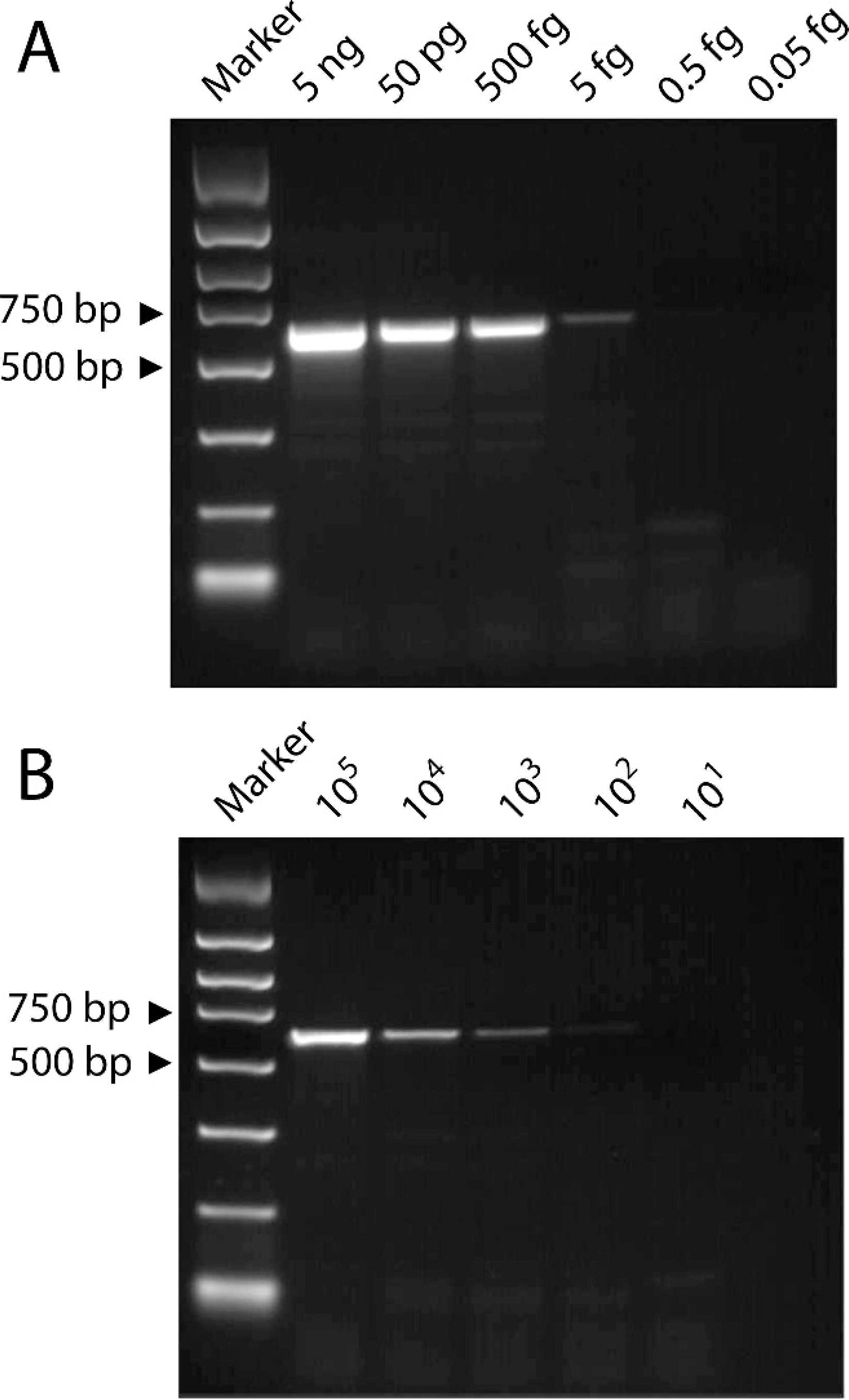
Detection limit of the polymerase chain reaction assay by using (
The PCR method described herein detected quantities of G. destructans gDNA as low as 5 fg. The lower limit of detection for conidia was 100 per 2 mg of wing skin. Sensitivity of conidia detection on skin may be influenced by resistance of the conidia to lysis, loss of fungal DNA during the extraction process, or partial inhibition of the PCR by substances in the wing tissue. However, histology demonstrated that the wings of bats infected with WNS were also colonized by fungal hyphae, which may enhance the ability to detect G. destructans in natural infections compared with spiked samples that contained only conidia.
Although PCR proved a more reliable method than culture, it was not as sensitive at detecting WNS as histology. The disparity may result from differences in the total coverage of wing area examined by each method. Only a single 3 × 3-mm piece of wing skin was used for PCR, which represented a relatively small proportion of the wing. In contrast, several sections cut from 1.5 × 3.0-cm rolls of wing tissue were used for histologic examination of each bat, which facilitated analysis of a greater proportion of the wing. Infections with a limited number of fungal foci are more likely to result in a false-negative result by PCR because of the small proportion of wing sampled. This could be addressed by conducting PCR analyses by using multiple skin samples from each animal. In addition, analyzing several bats from a suspected WNS-infested hiber-naculum increases the likelihood of detecting G. destructans if it is present.
The diagnostic specificity of PCR was 100% compared with histology. This strongly suggests that detection of G. destructans on wing tissue by PCR is synonymous with WNS infection. Polymerase chain reaction analysis has great utility as a rapid screening tool, and PCR followed by histology can be used to identify G. destructans as the causative agent of a fungal infection. If PCR alone is used as a surveillance tool, then follow-up sequencing of amplicons may be warranted.
For the current study, PCR primers were not tested for specificity against fungal isolates other than G. destructans. Although culture analyses demonstrated a diversity of fungi on bat wing skin, the absence of false-positive PCR results indicated that significant nonspecific amplification did not occur under the conditions used. Other studies underway in the authors' laboratory demonstrated that the primers used for the identification of G. destructans in wing skin did cross-react with other species of fungi found in cave sediments that were closely related to, but distinct from, G. destructans. Many of the bat carcasses analyzed for the current study were collected from cave floors where they were in direct contact with cave sediments and presumably fungi other than G. destructans, yet wing skin from these specimens did not produce false-positive results. In addition, sequencing analyses of the 624-nt amplification products generated from skin samples identified as PCR-positive were 100% identical to G. destructans in all instances. Therefore, there is strong evidence indicating that, although the primers may lack specificity for detecting G. destructans in environmental samples, they are highly specific for detection of G. destructans on bat wing skin.
Diagnosis of WNS in living bats is usually not feasible by using histology and/or culture techniques because of the relatively large amounts of tissue required. However, PCR could be used as a nonlethal technique to diagnose the disease by using wing-skin biopsy punches. 24 Such a technique would allow researchers to collect large numbers of samples to determine and track the prevalence of WNS over time, to facilitate nonlethal disease monitoring among endangered bat species, and to provide a means to assess the efficacy of potential treatments for WNS.
The emergence of WNS in the United States has caused great concern over the future of North American bat populations. As researchers and field biologists attempt to expand the knowledge base and monitor the spread of WNS, there is a pressing need for a rapid, economical, and accurate method to diagnose the disease. Methods such as histology and fungal culture have long turnaround times and require large amounts of tissue and specialized training to interpret; culture has low diagnostic sensitivity. Polymerase chain reaction requires only small amounts of tissue and offers a rapid, accurate, and economical alternative for diagnosis of WNS.
Acknowledgements
The authors thank P. Cryan (USGS) and H. Ip (USGS) for providing thoughtful reviews of the manuscript, numerous U.S. Fish and Wildlife Service personnel and state biologists for collecting and submitting bat carcasses, and A. Ballman (USGS) and K. Schuler (USGS) for coordinating sample collections. The authors also thank D. Berndt (USGS) for assisting with histology. This project was funded by the U.S. Geological Survey and by the U.S. Fish and Wildlife Service (intergovernmental agreement 501819H057). Use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Andrea Gargas and Carol Uphoff Meteyer contributed equally to this study.
Footnotes
a.
BD Diagnostic Systems, Sparks, MD.
b.
Gentra® Puregene® Genomic DNA Purification Kit, Qiagen Inc., Valencia, CA.
c.
NanoDrop 2000, Thermo Fisher Scientific Inc., Waltham, MA.
d.
OmniPrep™ for Fungi Kit, G-Biosciences, Maryland Height, MO.
e.
Quant-iT™ High-Sensitivity DNA Assay Kit, Invitrogen, Carlsbad, CA.
f.
GoTaq® Flexi, Promega Corp., Madison, WI.
g.
GelRed™, Phenix Research Products, Candler, NC.
h.
Sigma-Aldrich, St. Louis, MO.
i.
BigDye Terminator® v3.1, Applied Biosystems 3730xl DNA Analyzer, Applied Biosystems, Foster City, CA.
j.
Lasergene 5.0, DNASTAR, Madison, WI.