
Abstract
As concerns over the global spread of highly pathogenic avian influenza H5N1 have heightened, more countries are faced with increased surveillance efforts and incident response planning for handling a potential outbreak. The incorporation of molecular techniques in most diagnostic laboratories has enabled fast and efficient testing of many agents of concern, including avian influenza. However, the need for high-throughput testing remains. In this study, the use of a 384–well format for high-throughput real-time reverse transcription polymerase chain reaction (real-time RT-PCR) testing for avian influenza is described. The analytical sensitivity of a real-time RT-PCR assay for avian influenza virus matrix gene with the use of both 96– and 384–well assay formats and serial dilutions of transcribed control RNA were comparable, resulting in similar limits of detection. Of 28 hunter-collected cloacal swabs that were positive by virus isolation, 26 (92.9%) and 27 (96.4%) were positive in the 96– and 384–well assays, respectively; of the 340 hunter-collected swabs that were negative by virus isolation, 45 (13.2%) and 23 (6.8%) were positive in the 96– and 384–well assays, respectively. The data presented herein supports the utility of the 384–well format in the event of an avian influenza outbreak for high-throughput real-time RT-PCR testing.
Keywords
Most experts agree that an important aspect in the control and elimination of infectious disease outbreaks is the ability to provide quick and reliable diagnostic testing. As was seen recently in the United States, a key factor in the control of diseases with substantial economic consequences, such as exotic Newcastle disease (END) and highly pathogenic avian influenza (HPAI), was the ability to identify the causative agent quickly. 4,7 Because of the current situation with H5N1 Avian influenza virus (AIV) in Asia and Africa, concerns over the possibility of the next influenza pandemic have heightened; thus, surveillance efforts have escalated around the world, increasing testing demands on regional diagnostic laboratories.
For diagnostic laboratories to meet the demand for high-throughput testing needed in the face of an outbreak, as well as increased numbers of samples for surveillance, alternative methods need to be explored. The increased acceptance and wide use of molecular techniques in the diagnostic laboratory setting have resulted in improved and faster diagnostic tests, particularly with regard to high-consequence diseases, such as AI and END. With the development and approval of magnetic and glass bead nucleic acid extraction methods, the simultaneous extraction of up to 96 samples at one time is now possible, reducing time associated with manual extraction processes. 4,11 The 96–well platform for real-time reverse transcription polymerase chain reaction (real-time RT-PCR) has already been incorporated and validated for use with END and has applications with AI 4 ; however, processing a large number of samples during an outbreak (>2,000/day) would require several 96–well thermocyclers to meet the demands for fast turnaround times. Incorporation of the 384–well platform would require fewer thermocyclers and could test the equivalent of 4, 96–well plates in a single run, thereby increasing the number of samples tested at one time, hence reducing the turnaround time.
During the 2002 END outbreak in California, methods were developed in the midst of the outbreak to accommodate the testing demands. 4 Having faced this crisis, the need for equivalency testing and validation of methods that allow more rapid processing and testing of a larger number of samples is imperative. In this study, the feasibility of incorporating a 384–well-format real-time RT-PCR for increased throughput into the testing regime of the diagnostic laboratory, particularly for use in the event of an AI outbreak, is explored. The objectives of the study were 1) to evaluate the sensitivity and specificity of AI matrix gene real-time RT-PCR screening in a 96-well format compared with a 384–well format and 2) to compare inter- and intratest variability for the 96– and the 384–well formats.
The analytical sensitivities of the 2 real-time RT-PCR platforms were determined by using serial 10-fold dilutions (10–1–10–11) of in vitro transcribed AI matrix gene RNA, a which was then tested in triplicate. The real-time RT-PCR in the 96-well format was performed according to the protocol approved by the National Veterinary Services Laboratories (NVSL, Ames, IA) and used by the National Animal Health Laboratory Network (NAHLN) for screening avian samples for type A influenza virus. 8,9 Briefly, the test was performed with a 25–μl final reaction volume, a commercial RT-PCR kit b with AIV primers c and probe, d , 9 and a thermocycler e with threshold cycle (Ct) < 45, which was considered positive. Template RNA (1 μl) was added to each reaction, which was the only exception to the published protocol. The 384–well test was performed with a commercial AIV matrix gene reagent kit, b a 1–step real-time RT-PCR for the detection of AI matrix gene RNA with the use of 1 μl of template RNA in a 15–μl final reaction volume, and a real-time thermocycler, e as per the manufacturer's instructions, with the exception that Ct < 45 was considered positive (manufacturer recommendation is Ct < 40), for consistency with the NVSL protocol. Simple linear regression was performed to determine the analytical sensitivity of the 2 assays.
To evaluate the application of the 96– and 384–well assays to field samples, 368 cloacal swabs collected from hunter-harvested waterfowl during the 2007–2008 Texas hunting season were used (species included Blue- and Green-winged Teal, Northern Shoveler, and American Wigeon). Swabs were placed in 3.0–ml tryptose phosphate broth (TPB) f supplemented with antibiotics (penicillin G [2–103 U/ml], streptomycin [200 μg/ml], gentamicin [250 μg/ml], and amphotericin B [2–103 U/ml]) g in the field within 6 hr of harvest and were transported to the laboratory on ice where they were stored at −80°C until tested.
Virus isolation was performed on 368 cloacal swabs according to standard procedures. 10 Briefly, 0.2 ml of swab fluid was inoculated via the chorioallantoic sac route into 4, 9–10–day-old embryonating chicken eggs. The eggs were incubated at 37°C for 5 days and examined daily for embryo mortality. Any eggs with dead or dying embryos within 24 hr of inoculation were discarded as nonspecific. Amnio-allantoic fluid (AAF) was collected from all eggs with dead or dying embryos after 24 hr, and the fluids were tested for hemagglutinating (HA) activity. Eggs with surviving embryos 5 days postinoculation were chilled, and AAF was collected and tested for HA activity. Hemagglutination-positive fluids were further analyzed for the presence of AIV by real-time RT-PCR and a lateral flow antigen detection assay. h Fluids testing positive for AIV were submitted to the NVSL for confirmation and subtyping by hemagglutination inhibition (HI) and neuraminidase inhibition (NI) tests.
Fifty microliters of swab fluid (368 cloacal swabs) was dispensed into 96-well plates, and RNA was extracted with a commercial RNA isolation kit b according to the manufacturer's instructions in a magnetic particle processor. i Samples were tested for the presence of AI matrix gene with the use of a real-time RT-PCR in a 96- and a 384-well format. The 96- and 384-well tests were performed as listed above with the following exceptions: 8 μl and 5 μl, respectively, of template RNA was used, and for statistical analyses, Ct < 40 was considered positive. Samples that yielded values between 40.1 and 45 or produced no signal were classified as negative or “not determined.” A cutoff value of 40 was selected because of results obtained with the RNA dilution series (analytical sensitivity), the manufacturer's recommendations, b , e and the authors’ personal experience of not successfully isolating AIV from any samples that yielded Ct > 35 (>4,500 samples tested; data not shown). An RNA extraction control, 2 negative controls (extraction buffer), and an RNA-positive control provided with the kit were included with each 96-well extraction plate. a An internal control, Xeno, b was used with the AIV matrix gene reagent kit b in the real-time RT-PCR reaction to control for inhibition of the RT-PCR reaction.
Sensitivity and specificity, when using field samples, were calculated with the use of virus isolation as the reference test and a Ct value cutoff of 40 for the 96- and 384-well platforms. Exact binomial 95% confidence intervals (CIs) were calculated for the prevalence, sensitivities, and specificities. Exact McNemar tests were used to compare sensitivities and specificities between the 2 platforms. The estimated agreement between the 2 platforms was assessed with the kappa value. 6
Assay reproducibility and repeatability were assessed by comparing Ct values between both platforms with 30 cloacal swab samples (29 positive and 1 negative as determined by real-time RT-PCR for AI matrix gene) and an AI matrix positive extraction control. a Samples and positive control were extracted in triplicate and eluted in 85 μl of elution buffer. Eluates were pooled by sample to have a homogeneous solution with enough volume to conduct all repetitions. Samples were tested with the use of each platform twice per day on 2 different days to assess variability of samples tested in different plates and on different days. RNA samples were kept on ice between tests and frozen at −20°C between days. Linear mixed-effects models were used to compare the mean Ct value between the 96- and 384-well platforms for all samples with known Ct value. Threshold cycle value was modeled as a dependent variable, and random-effect terms were included for sample, day, and plate to account for dependence caused by analysis of replicate samples and variability attributable to unmeasured day and plate factors. Assay platform was included as a fixed-effect indicator variable, with the 96-well assay serving as the reference platform. Significance of the differences in means between the 96- and 384-well platforms was determined at P < 0.05. The proportion of variability in Ct value attributable to sample, day, and plate effects was estimated by dividing the respective variance component by the sum of random-effect and residual variances. Coefficients of variation (COV) were calculated by dividing the standard deviation by the mean and multiplying by 100. All statistical analyses were performed with available software. j
The analytical sensitivity for the 96- and 384-well platforms was comparable; the limit of detection for each assay was 10–8, which is approximately equal to 10 gene copies (Table 1). Regression analysis demonstrated linearity
Comparison of analytical sensitivity between the 2 real-time reverse transcription polymerase chain reaction platforms.
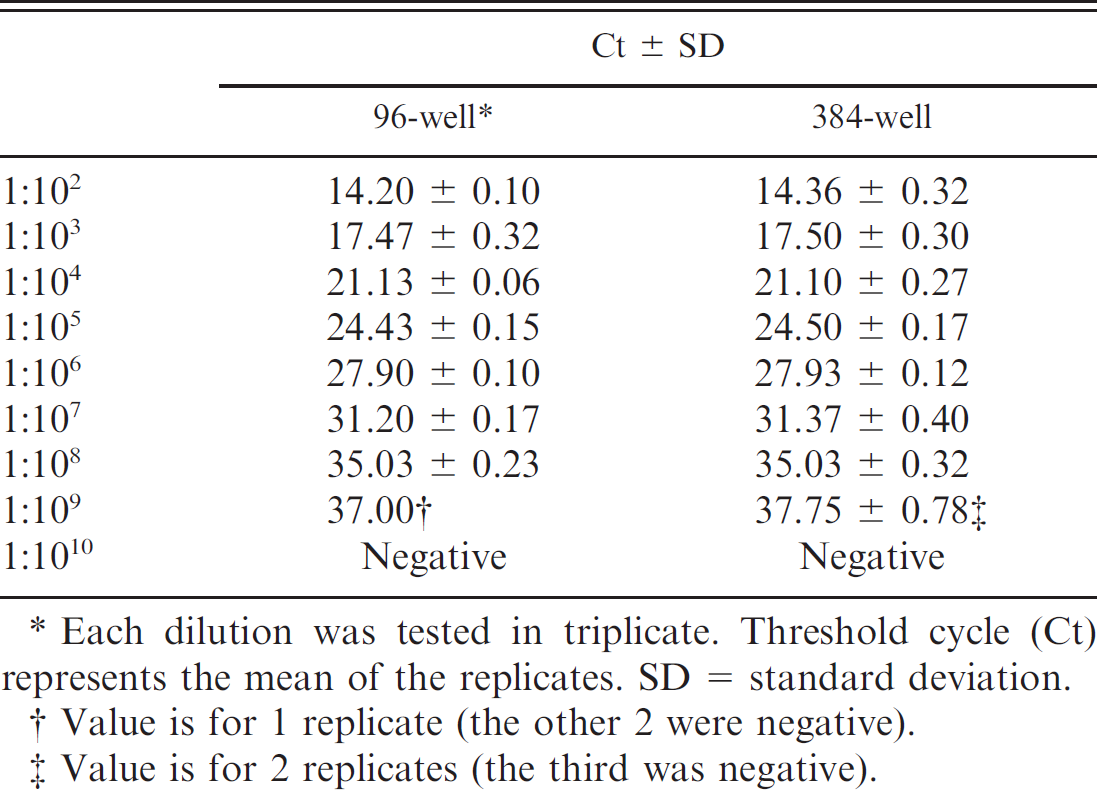
Each dilution was tested in triplicate. Threshold cycle (Ct) represents the mean of the replicates. SD = standard deviation.
Value is for 1 replicate (the other 2 were negative).
Value is for 2 replicates (the third was negative).
(r 2 = 0.9981 for both assays) and similar slopes (–3.405 + 0.03 [95% CI: −3.473 to −3.336] and −3.411 + 0.03 [95% CI: −3.478 to −3.344]) for 96– and 384–well assays, respectively) for both platforms.
The apparent prevalence of AIV in cloacal swabs of hunter-harvested waterfowl, as determined by virus isolation, was 7.6% (95% CI: 5.1–10.8), with a total of 28 samples positive for AIV. Of the 368 samples tested, 26 were positive and 295 negative for AIV by both the 96-well real-time RT-PCR assay and virus isolation (Table 2). In the 384-well platform, of the 368 samples tested, 27 were positive and 317 negative for AIV by both real-time RT-PCR and virus isolation (Table 2). The diagnostic sensitivity and specificity for the 96-well real-time RT-PCR platform, with virus isolation as the reference test, were 92.9% (95% CI: 76.5–99.1) and 86.8% (95% CI: 82.7–90.2), respectively. The diagnostic sensitivity and specificity of the 384-well real-time RT-PCR platform relative to virus isolation were 96.4% (95% CI: 81.7–99.9) and 93.2% (95% CI: 90.0–95.7), respectively. The difference in sensitivity between the 2 real-time RT-PCR assays was not significant (P = 1.00); however, the specificity of the 384-well format was higher (P < 0.0002) than for the 96-well
Summary of real-time reverse transcription polymerase chain reaction (real-time RT-PCR) platforms and virus isolation for individual samples tested for type A Avian influenza virus.
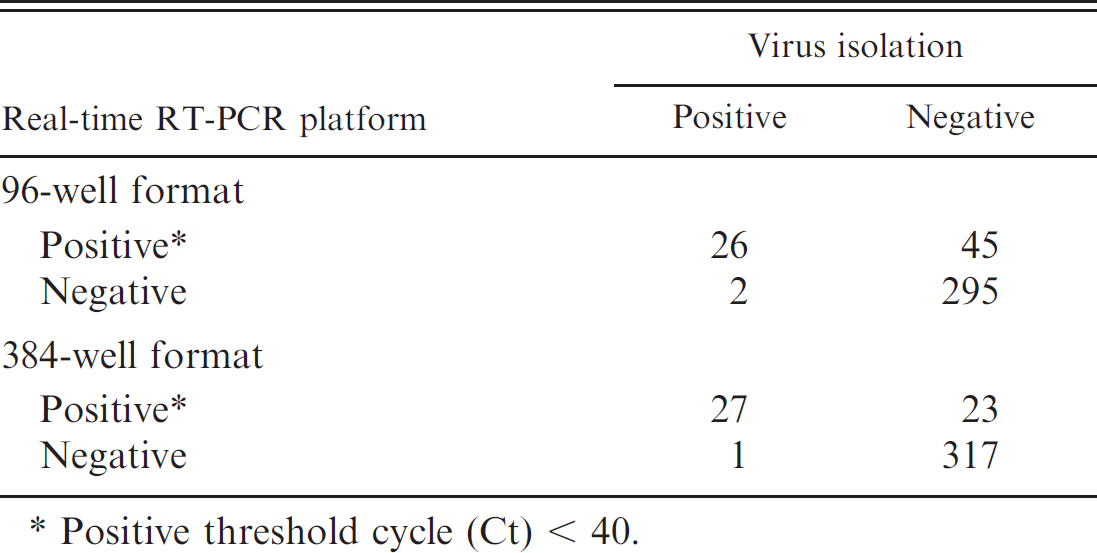
Positive threshold cycle (Ct) < 40.
Agreement of real-time reverse transcription polymerase chain reaction threshold cycle (Ct) values between the 96- and 384-well format for individual samples tested for type A avian influenza virus.
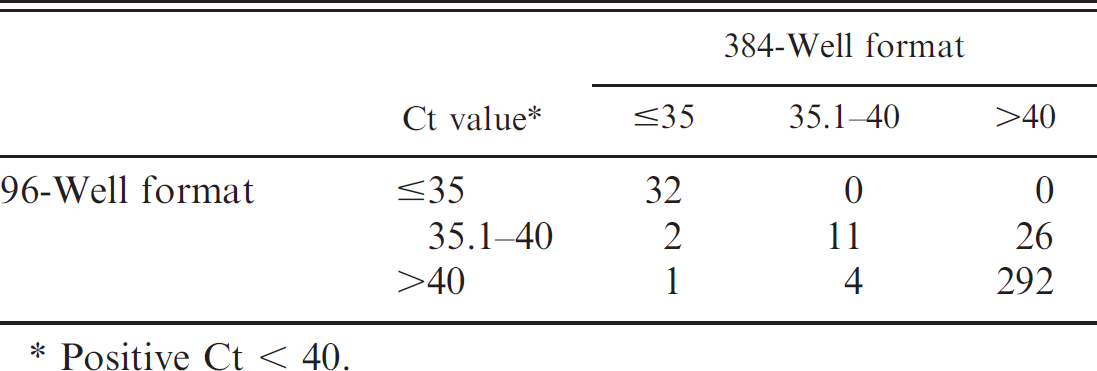
Positive Ct < 40.
format. Overall there was substantial agreement beyond chance between the 96- and 384-well assays; kappa = 0.711 (95% CI: 0.613–0.809). Of the discrepant results between the 2 real-time RT-PCR assays, all had Ct > 36 (Table 3), with the exception of 1 sample that was virus isolation-positive and positive with the 384-well assay (Ct of 25) but negative with the 96-well assay.
When known PCR-positive and -negative cloacal swab samples from hunter-harvested waterfowl were tested in triplicate, twice a day over 2 days, the mean Ct values were significantly lower (P < 0.001) for samples tested on the 384-well platform compared with those on the 96-well assay. Although significant, the difference was very small (0.45Ct; 95% CI: 0.53–0.36). Threshold cycle values for sample replicates were very similar (Fig. 1), and 98.8% of the model variance was attributed to the variance component for the random effect associated with sample identity. Unmeasured day and plate effects, summarized over both platforms, accounted for 0.1% and 0.3%, respectively, of the model variance. The coefficients of variation for the 96- and 384-well assays ranged from 0.01 to 7.23 and 0.06 to 7.8, respectively (data not shown).
In the case of an AI outbreak (HPAI or low-pathogenicity AI [LPAI]), thousands of samples might need to be tested daily, which would require high-throughput capability. In this report, the utility of a 384-well real-time RT-PCR platform versus virus isolation and the currently approved 96-well format for high-throughput sample testing was evaluated. The samples used in the study were cloacal swabs collected from wild waterfowl as part of the Avian Influenza Coordinated Agricultural Project (AI-CAP) Cooperative State Research, Education, and Extension Service (CSREES) AIV surveillance program. The apparent prevalence observed was low (7.6%) but consistent with previous reports. 2,5 The analytical sensitivity of the 2 assays, when using serial dilutions of transcribed control RNA, were comparable, resulting in similar limits of detection.
With the use of field samples, the sensitivity of our 96-well assay, relative to virus isolation, was higher (92.9%) than the sensitivity we calculated from Spackman et al. data 9 on an individual sample basis (66.6%), whereas the specificity of our assay was lower (87.4% vs. 94.9%). 9 One reason for this difference could be that some of the positive samples identified by the 96-well format and not detected by virus isolation could be “true positives” because the quality of the specimen can affect recovery in embryonated
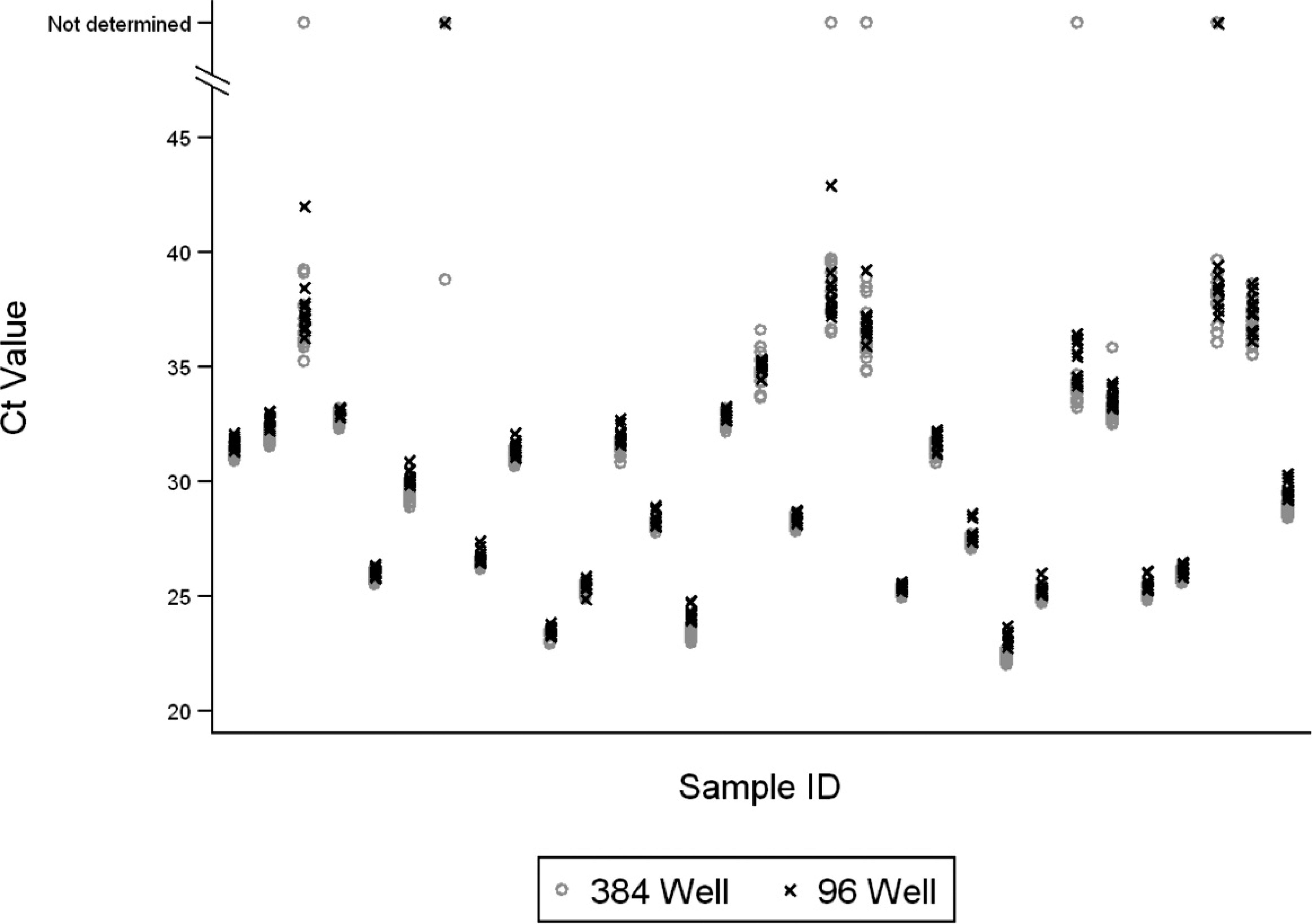
Observed threshold cycle (Ct) values from 96- and 384-well diagnostic platforms. Tests were classified as positive if the Ct value was <40 for both assays. Ct values for samples without a detectable signal after 45 cycles were classified as “not determined.”
chicken eggs but might not affect detection of the viral genome. However, we believe this issue is much more complex and other factors, such as the inability of certain viruses to grow in embryonated chicken eggs, presence of neutralizing antibodies in the specimen, or presence of inactivated viruses in the host itself, could affect virus recovery by virus isolation. Another reason for the differences observed in this study compared with that of Spackman et al. 9 could be the different RNA isolation methods used (magnetic beads vs. spin columns). In addition, the samples used in each study–-pools of up to 5 cloacal, tracheal, or environmental swabs from chickens and cloacal swabs from a live bird market in the Spackman et al. report 9 compared with individual cloacal swabs from hunter-harvested wild waterfowl in our study–-could have contributed to the differences observed.
Although differences were observed between the 2 realtime RT-PCR platforms evaluated here, the differences were not significant. The differences in sensitivity and specificity could be because of the use of virus isolation as the reference test in calculating sensitivity and specificity, in that virus isolation might not detect all viable viruses and does not detect inactivated viruses. Another possibility could be the different master mix used for the 2 assays. However, the base kit (AgPath-IDTM) was identical, with the only difference being that the kit used for the 384-well assay had the primers and probe included in the core master mix (AgPath-IDTM AIV-M Reagent kit b ), whereas in the kit used with the 96-well platform (AgPath-IDTM RT-PCR kit b ), primers and probe had to be added and were ordered from a different source. d
The data presented herein indicate very consistent results between the 96- and 384-well assays. Variance components for unmeasured day and plate effects accounted for less than 2% of total model variance when summarized across both platforms. Intercept-only models that were developed independently for the 96- and 384-well assays also attributed 98% of the model variance to random effects associated with sample identity (data not shown), indicating that both platforms are highly reproducible and robust with respect to variation between plates and days. Although the mean Ct value was significantly lower for samples tested in the 384-well format compared with the 96-well assay, the difference was 0.45Ct, which would generally not be considered biologically relevant when comparing real-time PCR results (usually a 3Ct difference, which is approximately equal to 1 log of virus, would be considered relevant). Overall, the 2 platforms performed equally well, and the 384-well format will likely be a practical and valid alternative to the 96-well format when a high number of samples need to be analyzed.
As was demonstrated during the END outbreak, by validating a 96-well RNA extraction platform, as well as 96-well format for real-time RT-PCR testing, instead of using the previously approved single-tube extraction and testing method, the number of samples processed daily increased from 184 to 1,900. 4 If the 384-well system were adopted, the number of samples that could be tested in 1 thermocycler would quadruple. An additional benefit would be a decrease in the cost of reagents because of the ability to test more samples in the 384-well format compared with the 96-well format with a reduced reaction volume: 15 μl (384-well) compared with 25 μl (96-well).
The combined use of the 96-well magnetic bead RNA extraction format with the 384-well platform for real-time RT-PCR allows the testing of 380 samples (with 4 wells for assay controls) within 3 hr, whereas the 96-well platform can test only 92 samples in the same amount of time, and corroborates the usefulness of a 384-well format. 1,3 One concern with the 384-well format could be potential cross contamination because of the small size of the wells. However, the reduced reaction volumes used in this study, together with the implementation of robotics and automation, could provide consistency in testing and would not be as influenced by fatigue or other human error.
In this study, the 384-well format was shown to perform as well as or better than the 96-well format as measured by sensitivity and specificity of the 2 real-time RT-PCR methods. Because virus isolation is not a perfect reference test with 100% sensitivity and specificity, the differences in specificities of the 96- and 384-well platforms might be explained in part by differences in how the 2 real-time RT-PCR assays performed on samples misclassified by virus isolation. More testing is needed for the validation of the 384-well platform; however, the data presented in the current study indicate that the 384-well format is a logical option for high-throughput testing, particularly in the midst of an outbreak, in which the prevalence (number of positive samples) and the viral load would be higher than in this study.
Acknowledgements. This project was partially funded by the CSREES AICAP “Prevention and Control of Avian Influenza in the US” grant awarded to B. Lupiani. The authors thank Feng Sun for valuable technical assistance and Dr. Alfonso Clavijo for critically reviewing the manuscript.
Footnotes
a.
National Veterinary Services Laboratories, Ames, IA.
b.
AgPath-IDTM RT-PCR kit, AgPath-IDTM AIV-M Reagent kit, MagMaxTM-96 Viral RNA Isolation kit; Applied Biosystems, Foster City, CA.
c.
Sigma-Genosys, The Woodlands, TX.
d.
Biosearch Technologies Inc., Novato, CA.
e.
ABI 7500 Fast (96-well), ABI 7900HT (384-well); Applied Biosystems, Foster City, CA.
f.
BD, Franklin Lakes, NJ.
g.
Sigma-Aldrich, St. Louis, MO.
h.
FluDetectr kit, Synbiotics Corp., San Diego, CA.
i.
KingFisher 96 magnetic particle processor, Thermo Fisher Scientific Inc., Waltham, MA.
j.
Intercooled Stata 9.2 for Windows, StataCorp LP, College Station, TX.