
Abstract
Cytauxzoon felis infection in domestic cats has historically been nearly 100% fatal. However, increasing reports of domestic cats that survive cytauxzoonosis and reports of asymptomatic cats with C. felis infections suggest the existence of different parasite strains that vary in pathogenicity. The objective of the current study was to obtain epidemiologic information about cytauxzoonosis through genotypic characterization of archived histologic specimens from domestic cats with C. felis infections that were diagnosed in Georgia between 1995 and 2007. Such retrospective data on genetic variability will provide an historic context for current studies of C. felis genotype frequencies. Cytauxzoon felis DNA was obtained from formalin-fixed, paraffin-embedded tissues from infected cats diagnosed with cytauxzoonosis at necropsy. Genetic characterization of C. felis was performed using sequence analysis of the polymerase chain reaction–amplified ribosomal internal transcribed spacer regions 1 and 2 (ITS1, ITS2). Eleven different combined ITS1 and ITS2 sequences were identified, the majority of which were identical to those previously reported in fatally infected cats from Georgia. The findings of the current study document the existence of genetically distinct C. felis populations in historical samples and, together with data from contemporary samples, demonstrate a diverse population structure for C. felis.
Introduction
Fatal cytauxzoonosis was first described in a domestic cat from Missouri in 1976. 28 Since that time, Cytauxzoon felis (family Theileriidae) infections in domestic cats have been detected throughout the southeastern, south-central, and mid-Atlantic regions of the United States. Experimental infections have shown both Dermacentor variabilis 3 and Amblyomma americanum 25 to be competent vectors of the parasite. The bobcat (Lynx rufus) is believed to be the reservoir host of this parasite 3,11 ; however, asymptomatic infection also has been detected in other wild felids in the United States, including panthers and cougars (Puma concolor coryi, P. c. stanleyana, and P. c. couguar). 16,27,30 Infection with Cytauxzoon spp. has also been identified in felids in other parts of the world, as well. C. felis infection has been reported in ocelots, 1 pumas, 1 jaguars, 1 and captive-reared lions 23 in Brazil, in addition to the identification of piroplasms morphologically indistinguishable from Cytauxzoon spp. in domestic cats. 20 In Europe, domestic cat and Iberian lynx infection with Cytauxzoon spp. have been reported from Spain, 8,21 as well as a single domestic cat infection from France. 7 In Asia, similar intraerythrocytic piroplasms with the proposed name Cytauxzoon manul have been detected from Pallas's cats in Mongolia. 26
In contrast to the common asymptomatic infection of bobcats and cougars in the United States, C. felis infection of domestic cats usually results in rapid and severe disease progression, and most infected cats die within 1 week of initial clinical illness. 13,17 However, more recently published reports have identified cats that have survived C. felis infection. 6,12,19,22,29 Supportive care and antiprotozoal drug administration have had inconsistent therapeutic results and limited success in treating cytauxzoonosis. 2,12,13,22 Furthermore, some cats have survived both natural and experimental C. felis infections without medical treatment. 6,19,22,29 Thus, treatment protocol is an unlikely explanation for the survival of these individuals. It is speculated that surviving cats may have recovered because they were infected with a less virulent strain of C. felis..
Currently, very little is known about the C. felis genome and only sequences for the ribosomal genes have been characterized and submitted to GenBank. One previous study 19 compared sequenced portions of the C. felis 18S ribosomal RNA (rRNA) gene between 2 infected cats and found that the sequences were virtually identical. A second study 4 comparing C. felis sequence data obtained from 3 infected ticks and 1 naturally infected cat reported >99.8% sequence similarity between each sequence and the previously published sequences. 19 These findings are not unexpected because the 18S region is a highly conserved area of the genome encoding a functional RNA molecule. In contrast, the internal transcribed spacer (ITS) regions are noncoding regions that do not have the structure-function constraints of the rRNA genes. As such, the ITS regions are likely to evolve more quickly and are more likely to show variability among putative strains of C. felis. The earlier study 4 comparing C. felis sequence data from infected ticks also analyzed data for the C. felis first ITS region (ITS1). Two polymorphic bases were found within 1 sample when comparing ITS1 sequence data from 2 infected ticks and 1 naturally infected cat. Otherwise, the sequences within the ITS1 region were identical. A more recent and comprehensive study 5 of the first and second ITS regions of the C. felis rRNA operon have detected unique C. felis ITS1 and ITS2 sequences from naturally infected cats in Arkansas and Georgia. The unique ITS1 and ITS2 sequences described in that study varied geographically and had a significant association with clinical outcome (survival vs. fatal infection) in infected domestic cats. The objective of the current study was to characterize the C. felis genome via sequencing of archived paraffin-embedded tissue samples to provide an important historic context for current studies of C. felis genotype frequencies in infected domestic cats.
Materials and methods
Samples
A retrospective, computer-generated search was initiated using archived medical records at The University of Georgia (UGA) Veterinary Diagnostic Laboratory (Athens, GA) and the Tifton Diagnostic & Investigational Laboratory (Tifton, GA) to identify domestic cats that were diagnosed at necropsy with cytauxzoonosis. For each individual, the pathologist's diagnosis of cytauxzoonosis was based on the presence of schizonts within phagocytic cells that were morphologically consistent with C. felis as identified on formalin-fixed, paraffin-embedded (FFPE), hematoxylin and eosin–stained necropsy tissues. Archived FFPE tissues were retrieved from 98 cats with C. felis infections. These diagnostic cases encompassed a time period from 1995 through 2007.
DNA extraction
DNA was isolated from archived FFPE tissues by shaving approximately 15 mg of material from the paraffin blocks. Spleen was preferentially used when present; otherwise, portions of lung, liver, or kidney were collected, and only 1 tissue sample was used per animal. The paraffin-embedded tissue samples were incubated with xylene overnight followed by a series of ethanol washes. DNA was extracted using a commercially available kit, a modified by a prolonged 3-day tissue digestion with an additional 20 μl proteinase K b added each day. Extracted DNA was eluted using a 1:2 dilution of the extraction kit's elution buffer in molecular biology grade water. After the elution process, the DNA was stored at 4°C until analyzed.
To prevent cross-contamination, new blades and tools were used for each specimen, and DNA extraction, amplification setup, and postamplification steps were each performed in a separate area of the laboratory with separate pipettes.
Polymerase chain reaction
Polymerase chain reaction (PCR) analysis was performed on all samples using forward and reverse oligonucleotide primers designed to anneal to conserved areas flanking the ITS1 and ITS2 regions of the C. felis genome. Design of the primer pairs was accomplished using sequence data from GenBank. The sequences of the forward and reverse oligonucleotide primers used to amplify C. felis ITS1 were 5′-CGATCGAGTGATCCGGTGAATTA-3′ and 5′-GCTGCGTCCTTCATCGATGTG-3′, respectively. These primers were expected to produce an amplicon of 651 bp from genomic C. felis DNA that incorporated the 458-bp ITS1 region plus 18S and 5.8S partial flanking regions. The sequences of the forward and reverse oligonucleotide primers used to amplify C. felis ITS2 were 5′-TGAACGTATTAGACACACCACCT-3′ and 5′-TCCTCCCGCTTCACTCGCCG-3′, respectively. These primers were expected to produce an amplicon of 431 bp from genomic C. felis DNA that incorporated the 265-bp ITS2 region plus 5.8S and 28S partial flanking regions.
For samples in which PCR was unable to amplify the entire C. felis ITS1 region, PCR analysis was repeated using an additional primer pair designed to produce a smaller, 290-bp amplicon that incorporated 155 bp of ITS1 plus the 18S partial flanking region. This shorter region within ITS1 was targeted based on increased variability previously detected within this region. 5 The sequences of the forward and reverse oligonucleotide primers used to amplify this region within C. felis ITS1 were 5′-ATAGAGTAAACGCTTCCTTCGGG-3′ and 5′-CGCAGAAGTCTGCAAGTCACAATG-3′, respectively.
Components of the PCR reaction mixture included a 2X hot start Taq polymerase master mix, c containing hot start DNA polymerase, PCR buffer with 3 mmol of MgCl 2 , and 400 μmol of each deoxyribonucleotide triphosphate (dNTP); additional MgCl 2 to bring the final MgCl 2 concentration to 2.0 mmol; 37.5 pmol of each primer; 4 μl of DNA template; and molecular biology grade water to adjust the reaction mixture to a final volume of 50 μl. One known-positive, whole blood–derived C. felis template and 2 negative templates (consisting of a known C. felis–negative feline blood sample and molecular biology grade water) were used as controls for all amplification reactions.
Nucleotide variation and frequency of Cytauxzoon felis internal transcribed spacer region (ITS1 and ITS2) sequences.
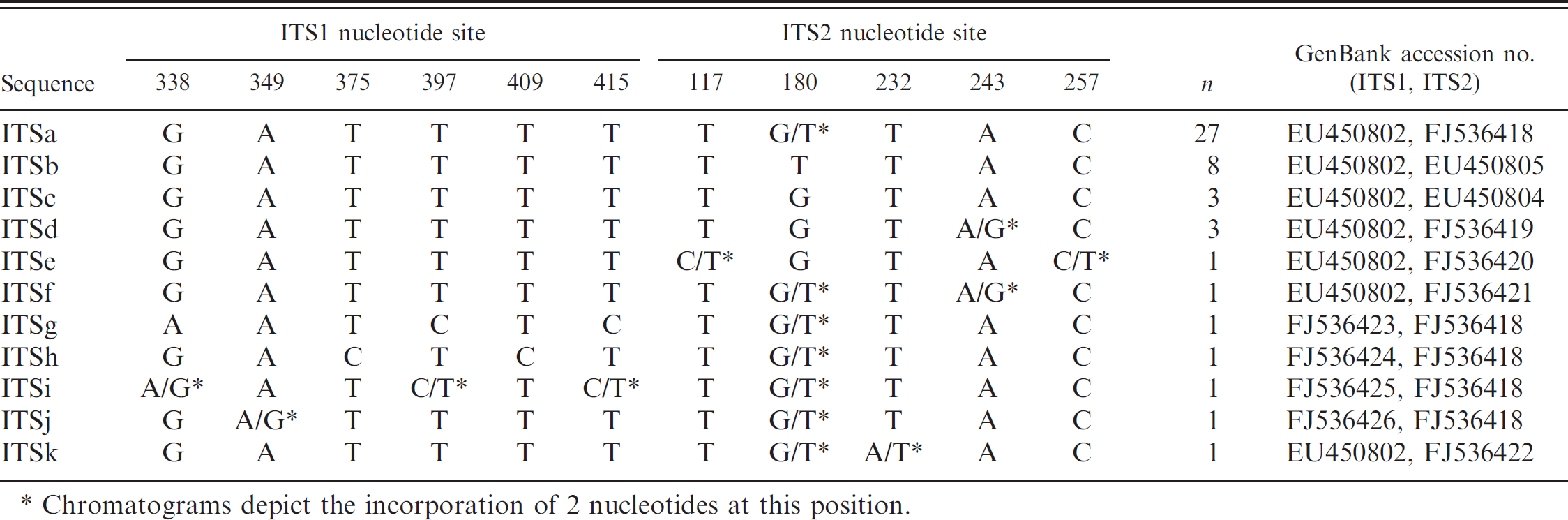
Chromatograms depict the incorporation of 2 nucleotides at this position.
Reaction mixtures were assembled in a PCR enclosure with an ultraviolet light d so that work surfaces could be decontaminated and trace DNA denatured in between reactions.
After initial denaturation at 95°C for 5 min, 38 amplification cycles were performed. Each cycle was comprised of 30 sec at 94°C, 30 sec at 60°C, and 1 min at 72°C, with final extension at 72°C for 10 min. The PCR products were resolved by electrophoresis (100 V, 30 min), using agarose gels that were prestained with ethidium bromide and visualized with ultraviolet light.
DNA sequencing and analysis
The PCR products producing single positive bands at the appropriate target lengths were purified using a commercial kit. e Automated sequencing was then performed at a commercial f or university g laboratory, using the forward and reverse primers from the PCR amplifications. Chromatogram data were carefully reviewed for each sample and ambiguous sequence data were excluded from further analysis. Forward and reverse sequence data were assembled and then aligned using Vector NTI software h to identify any polymorphisms within the ITS regions of the amplicons. Sequencing results were then analyzed chronologically to identify temporal relationships among different C. felis genotypes.
Results
C. felis ITS amplification and sequencing
Initial amplification of the C. felis ITS1 region from archived tissues of infected domestic cats yielded high-quality sequence data for 21 of 98 specimens, and amplification of the C. felis ITS2 region yielded high-quality ITS2 sequence data for 85 of 98 specimens. Repeat PCR analysis that restricted the length of the target area within ITS1 to a 290-bp region provided high-quality sequence data for an additional 27 specimens.
Analysis of the 48 combined C. felis ITS sequences (ITS1 and ITS2) revealed 6 single nucleotide polymorphisms (SNPs) within the restricted target region of ITS1 and 5 SNPs within ITS2 (Table 1). The most common ITS sequence, labeled here as ITSa and detected in 27 of 48 (56.3%) samples, contained one polymorphic site within ITS2 for which chromatograms revealed the incorporation of 2 different nucleotides at this single locus (herein referred to as mixed nucleotide positions). The ITSb sequence, detected in 8 of 48 (16.7%) samples, and the ITSc sequence, detected in 3 of 48 (6.3%) samples, had ITS1 nucleotide sequences identical to ITSa and differed only at the same polymorphic site (position 180) within ITS2, where each contained 1 of the 2 nucleotides incorporated at the mixed site in ITSa. The ITSg and ITSh sequences, detected in a single sample each, differed from ITSa at 3 SNPs and 2 SNPs, respectively, each at different loci within ITS1. The ITSd sequence was detected in 3 of 48 samples (6.3%) and the remaining ITS sequences (identified here as ITSe, ITSf, ITSi, ITSj, and ITSk) were detected in a single sample each. Each of these sequences differed from those previously described by the inclusion of mixed nucleotides at 2–4 positions, where 1 of the nucleotides in each case represented the more commonly incorporated nucleotide in the other alleles. All variable and mixed nucleotide SNPs were confirmed by repeating the amplification and sequencing reactions.
Chronologic distribution of C. felis ITS sequences
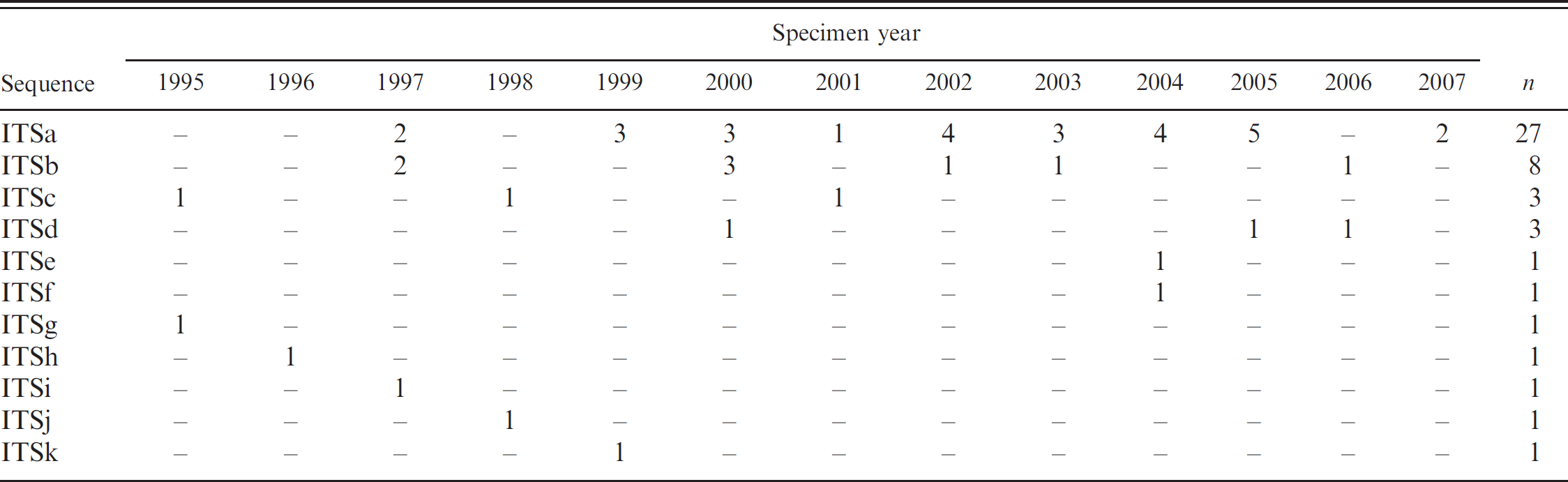
The majority of the different C. felis sequences were detected in a single sample from a single year and not detected again. Another 2 sequence types were identified in single samples from 3 different years. The remaining 2 sequence types, comprising the vast majority of samples, were detected in specimens scattered throughout the years studied, with no obvious temporal trends. The chronologic distribution of the C. felis ITS sequences is summarized in Table 2.
Chronologic distribution of Cytauxzoon felis internal transcribed spacer region (ITS1 and ITS2) sequences.
Discussion
Extraction and PCR amplification of DNA from FFPE tissues poses multiple challenges. Formalinfixation results in cross-linking of protein to nucleic acids, making DNA extraction difficult. In addition, extraction procedures commonly result in DNA fragmentation that limits the size of the target DNA available for PCR analysis. It has been demonstrated that, with increasing age of the specimen and time in formalin fixative, longer DNA fragments are amplified with decreasing success. 10,14,15 Hence, it is not surprising that amplification of the 651-bp region containing the complete ITS1 sequence was less successful than amplification of the shorter 431-bp region containing ITS2. Because DNA compromised by fixation and age may require an amplification strategy that produces smaller PCR products, an additional PCR amplification of a smaller region within ITS1 (290 bp) was performed to obtain additional sequence data for the present study. This shorter region was targeted to amplify and sequence 7 of the 9 variable nucleotide sites identified in an earlier study of C. felis ITS1 sequence variability in domestic cats with cytauxzoonosis. 5
The degree of sequence polymorphism seen in the current set of C. felis samples is similar to that which has been observed in previous studies. The majority of the C. felis ITS sequence types detected in the current study were identical to genotypes previously detected in domestic cat infections. 5 The C. felis ITS sequences unique to the current study were present predominantly in individual samples and differed at 1–3 nucleotides, a level of polymorphism comparable to that detected among all C. felis ITS1 and ITS2 sequences available in GenBank.
Detailed review of the sequence chromatograms revealed the incorporation of 2 nucleotides at single, variable positions within ITS1 or ITS2 in the majority of samples. This finding may reflect coinfection of the cat with multiple C. felis genotypes. Alternatively, because the DNA sequencing was based on a PCR pool, it could reflect polymorphisms in multiple copies of the rRNA genes that may exist in the C. felis genome, as described in some isolates of closely related parasites, such as Theileria parva 18 and some Babesia spp. 9,24 Resolution of this issue is difficult given the current lack of a cell culture system that would allow establishment of a clonal line of C. felis. Alternatively, the use of a Southern blot analysis may allow identification of multiple gene copies, if present.
For the chronologic analysis, there was year-to-year variability in the C. felis ITS sequences detected, though with no obvious temporal trend. The most common sequence, ITSa, was found in samples from infected cats scattered among the years analyzed and is identical to the sequence from a single isolate from an infected domestic cat in Georgia reported in a study of more contemporary isolates. 5 Similarly, the second most common ITS sequence, ITSb, was also found scattered throughout the years analyzed, and this sequence is identical to a common sequence in isolates from those more current cat infections in Georgia. 5 Interestingly, the most common C. felis ITS sequence described recently 5 in samples from 2005–2007 was identified in only 3 of the samples in the current study, and this sequence was not detected in samples since 2001. The significant difference in the ITS genotypes which are most commonly detected suggests that changes have occurred in the C. felis population over the time frame covered by the current and previous study. 5
It is interesting to compare the sequences from the current study to those identified previously as having a fatal or nonfatal outcome of infection in domestic cats. The most common ITS sequence in the current study (ITSa) was only detected in 1 sample in the previous study (sequence h), 5 and this cat was fatally infected. The next most common sequence identified in the necropsy cases of the current study (ITSb) was identical to a common genotype (ITSB), 5 which was significantly associated with a fatal outcome of infection in that study. Conversely, the common genotype in the previous study associated with nonfatal infection (ITSA) 5 was detected in only 3 of the 48 necropsy cases (6.3%) of the current study. Because the population in the current study included all cats that were diagnosed with cytauxzoonosis at necropsy, it is likely these samples represent isolates from both euthanized and fatally infected domestic cat infections. Because many factors affect an owner's and veterinarian's decision to euthanize an infected cat, it is impossible to make a more direct comparison of the 2 studies' sequencing results, which would require an assumption that those cats presenting to necropsy would have a fatal outcome of natural infection. Nonetheless, the C. felis ITS sequences associated with fatal outcomes in a previous study 5 were the sequences most commonly detected in the necropsy cases of the current study.
The sample population of the current study was limited to those archived tissues for C. felis–infected cats that were available from the State of Georgia, and a previous study 5 identified a much greater percent of fatally infected cats among those C. felis isolates from Georgia versus Arkansas. A more comprehensive genetic analysis comparing archived C. felis samples over a broader geographic area, including states with higher survival rates for infected cats, may better identify sequence diversity and trends expected with the possible emergence of less pathogenic strains of C. felis.
In summary, the current study examines the variability of C. felis ITS sequences from domestic cats in Georgia that were diagnosed with cytauxzoonosis at necropsy from 1995 to 2007. The identification of 11 different sequences lends further support to the existence of genetically distinct C. felis populations. The majority of the C. felis ITS sequences identified in the current study were identical to those previously reported in fatally infected cats from Georgia. Identification of other areas of the C. felis genome, ideally that encode proteins that affect pathogenicity, and a comparison of sequences from archived samples from infected cats in other geographic areas outside the State of Georgia may provide further support for the existence of different parasite strains that underlie the increased survival of more recently infected cats. Further disclosure of the genetic characterization of the C. felis population structure will contribute greatly to a better understanding of the epidemiology of cytauxzoonosis in domestic cats.
Acknowledgements
The authors wish to thank Drs. Pauline Rakich (Athens Veterinary Diagnostic Laboratory, Athens, GA) and Alan Liggett (Tifton Diagnostic & Investigational Laboratory, Tifton, GA) for their efforts in sample collection and submission. Technical assistance over the course of this study was provided by Mr. Andrew Allison, Dr. Elizabeth Howerth, Ms. Kristine Yu, and Mr. Jonathan Katz. Funding for this project was provided by the Morris Animal Foundation.
Footnotes
a.
QIAamp® DNA Mini-Kit, Qiagen Inc., Valencia, CA.
b.
Sigma-Aldrich, St. Louis, MO.
c.
HotStarTaq® Plus Master Mix, Qiagen Inc., Valencia, CA.
d.
Labconco Corporation, Kansas City, MO.
e.
QIAquick® PCR Purification Kit, Qiagen Inc., Valencia, CA.
f.
MWG Biotech Inc., High Point, NC.
g.
University of Georgia Integrated Biotechnology Laboratories, Athens, GA.
h.
Invitrogen Corp., Carlsbad, CA.