
Abstract
Although cytauxzoonosis has historically been nearly 100% fatal in domestic cats, increasing number of reports of infected cats that demonstrate less-severe disease suggest the existence of different strains of Cytauxzoon felis. To test this hypothesis, the genetic variability of C. felis was examined in blood samples from naturally infected domestic cats from Arkansas and Georgia by using the first and second ribosomal internal transcribed spacer regions (ITS1, ITS2) as markers to assess genotypic variability. In addition, the clinical outcome of infection (survival vs. fatal disease) was analyzed. Within the C. felis ITS1 region, there were a total of 8 single nucleotide polymorphisms (SNP) and a single nucleotide insertion. Within the ITS2 region, there were a total of 4 SNPs and a single 40 base pair insertion. When taken together, the ITS1 and ITS2 sequence data defined a total of 11 different sequences and 3 unique genotypes. One unique ITS1-ITS2 genotype was detected in samples submitted exclusively from Arkansas, and a second unique genotype was submitted exclusively from Georgia. There was a significant association between infection with C. felis that contained particular ITS genotypes and survival of the infected domestic cat. The identification of unique C. felis genotypes obtained from different geographic areas and the association of particular ITS genotypes with the outcome of infection suggest the existence of parasite strains that may vary in pathogenicity to the domestic cat and offer an explanation for the survival of some infected cats in more recent case studies.
Introduction
Cytauxzoonosis is an emerging, highly fatal, tick-borne, hemoprotozoal disease of domestic and exotic cats. The causative organism, Cytauxzoon felis (family Theileriidae, genus Cytauxzoon), is a relatively new pathogen in the United States that was first detected in Missouri in 1973 15 and subsequently throughout the southeastern, south-central, and mid Atlantic regions of the United States. The bobcat (Lynx rufus) was identified as the natural reservoir host of this parasite. 3,5 In affected domestic cats, the course of disease is usually rapid, and most cats die within 1 week of initial clinical illness. 7,9,17 The sporadic occurrence, short course of illness, and high fatality rate of cytauxzoonosis in domestic cats suggest that domestic cats likely serve as aberrant or dead-end hosts. 7,11
Although C. felis infection historically has been viewed as uniformly fatal in domestic cats, more recent studies document cats that have survived infection. 4,6,12,13,16 However, supportive care and antiprotozoal drugs have had inconsistent therapeutic results and limited success in treating cytauxzoonosis. 2,6,7,13 Furthermore, some cats that survived C. felis infections (both natural and experimental) received no therapy, 12,13,16 which suggests that treatment protocol is an unlikely explanation for the survival of these infected cats. Another explanation is that these cats recovered because they were infected with a less-virulent strain of C. felis. It is notable that the majority of these cats that survived cytauxzoonosis were documented in a relatively limited geographic area (the Midwest, including northwest Arkansas), which might be expected with the emergence of different genetic strains of the parasite.
An important step in assessing potential strain variation within the C. felis population is the use of a suitable gene as a genetic marker of variability. Currently, very little is known about the C. felis genome. Only sequences for the ribosomal genes have been characterized and submitted to GenBank. One previous study 12 compared sequenced portions of the C. felis ribosomal RNA (rRNA) gene from a single cat that survived infection with sequences reported in GenBank. All sequences were shown to be virtually identical. The selected genomic target in that study was the small subunit rRNA gene, which is a highly conserved region encoding a functional RNA molecule. In contrast, the first and second internal transcribed spacer regions of the rRNA operon (ITS1, ITS2) are noncoding regions and, thus, do not have the structure–function constraints of the rRNA genes. As such, the ITS regions are likely to evolve more quickly 8 and are more likely to show variability among putative strains of C. felis. Variability of the ITS regions has been recognized in other apicomplexan species, such as Babesia canis, Cyclospora cayetanensis, and Eimeria spp. 1,14,18 Three closely related species of Babesia canis that vary in pathogenicity for the dog share identical small subunit rRNA sequences but are individually distinguishable by variability in their ITS sequences. 18
Genetic analysis of the C. felis ITS regions from cats with cytauxzoonosis may allow identification of genetically distinct parasite populations and, if present, may offer a possible explanation for the survival of some infected cats in more recent case reports. The aims of the current study were to assess whether the ITS1 and ITS2 regions of ribosomal DNA (rDNA) could be used as genetic markers of distinct C. felis populations, including those from different geographic areas and those that vary in pathogenicity in the domestic cat, and to establish the foundation for further studies on molecular epidemiology and diagnostics of cytauxzoonosis.
Materials and methods
Parasite samples
Cytauxzoon felis–infected blood samples from acutely ill cats diagnosed with cytauxzoonosis from 2005 to 2007 were submitted from collaborating veterinary hospitals and diagnostic laboratories in Arkansas and Georgia. For each case, the pathologist's diagnosis of cytauxzoonosis was based on the presence of erythrocytic piroplasms that were morphologically consistent with C. felis as identified on stained peripheral blood smear review. Samples from Arkansas were submitted by the Veterinary Diagnostic Laboratory at the Arkansas Livestock and Poultry Commission (Little Rock, AR). Samples from Georgia were obtained from the University of Georgia (UGA) Veterinary Teaching Hospital (Athens, GA), the UGA Veterinary Diagnostic Laboratory (Athens, GA), and the UGA Veterinary Diagnostic and Investigational Laboratory (Tifton, GA). Follow-up telephone communication with submitting veterinarians confirmed the presence of clinical disease consistent with acute cytauxzoonosis at the time of the blood-sample submission, as well as identified the clinical outcome of acute infection for each cat.
DNA extraction and polymerase chain reaction amplification
DNA was extracted from ethylenediamine tetra-acetic acid anticoagulated whole blood samples by using a commercially available kit a according to manufacturer's instructions. Polymerase chain reaction (PCR) analysis was performed on all samples by using forward and reverse oligonucleotide primers designed to conserved regions that flank the ITS1 and ITS2 regions of C. felis. Primer pairs were designed by using sequence information from GenBank. The sequences of the forward and reverse oligonucleotide primers used to amplify C. felis ITS1 were 5′-CGATCGAGTGATCCGGTGAATTA-3′ and 5′-GCTGCGTCCTTCATCGATGTG-3′, respectively. These primers were expected to produce an amplicon of 651 base pair (bp) from genomic C. felis DNA that incorporates the 458 bp ITS1 region plus 18S and 5.8S partial flanking regions. The sequences of the forward and reverse oligonucleotide primers used to amplify C. felis ITS2 were 5′-TGAACGTATTAGACACACCACCT-3′ and 5′-TCCTCCCGCTTCACTCGCCG-3′, respectively. These primers were expected to produce an amplicon of 431 bp from genomic C. felis DNA that incorporates the 265 bp ITS2 region plus 5.8S and 28S partial flanking regions. The PCR components consisted of a 2× hot start Taq polymerase master mix b that contains hot start DNA polymerase, PCR buffer with 3 mmol of MgCl2, and 400 μmol of each deoxyribonucleotide triphosphate (dNTP); additional MgCl2 to bring the final MgCl2 concentration to 2.0 mmol; 37.5 pmol of each primer; 4 μl of DNA template; and molecular-biology-grade water to adjust the reaction mixture to a final volume of 50 μl. One known positive template and 2 negative templates (consisting of a known negative sample and molecular-biology-grade water) were used as controls for all amplification reactions.
After the initial denaturation at 95°C for 5 min, 35 amplification cycles were performed. Each cycle was composed of 30 sec at 94°C, 30 sec at 60°C, and 1 min at 72°C, with final extension at 72°C for 10 min. The PCR products were resolved by electrophoresis (100 V, 30 min) by using agarose gels that were prestained with ethidium bromide and subsequently visualized with ultraviolet light.
DNA sequencing and analysis
The PCR products were purified by using a commercial kit, c and automated sequencing was then performed at a commercial d or university e laboratory by using the forward and reverse primers used for PCR. Sequences and chromatogram data were carefully analyzed, and forward and reverse sequence data were assembled by using the Contig Express module of Vector NTI software. f Assembled sequences were then aligned by using Vector NTI software f to identify any polymorphisms within the ITS regions of the amplicons. Combined ITS1–ITS2 sequence data from each sample were evaluated with regard to state of origin (Arkansas vs. Georgia) to determine if the presence of particular parasite genotypes varied geographically. By using a chi-square test for independence, the unique C. felis ITS genotypes were evaluated with regard to clinical outcome (survival vs. fatal infection) to determine if there was any association between unique parasite genotypes and observed pathogenicity. All tests were performed by assuming a 2-sided alternative hypothesis, and P values <0.05 were considered statistically significant. When ≥1 cells in a contingency table had an expected count < 5, the Fisher exact test was used. Because many factors other than pathogenicity may affect an owner's decision to euthanize his or her cat, cases in which the infected cat was euthanized were excluded from the analysis.
. Nucleotide variation and frequency of Cytauxzoon felis first and second ribosomal internal transcribed spacer region (ITS1 and ITS2) genotypes and sequences.*
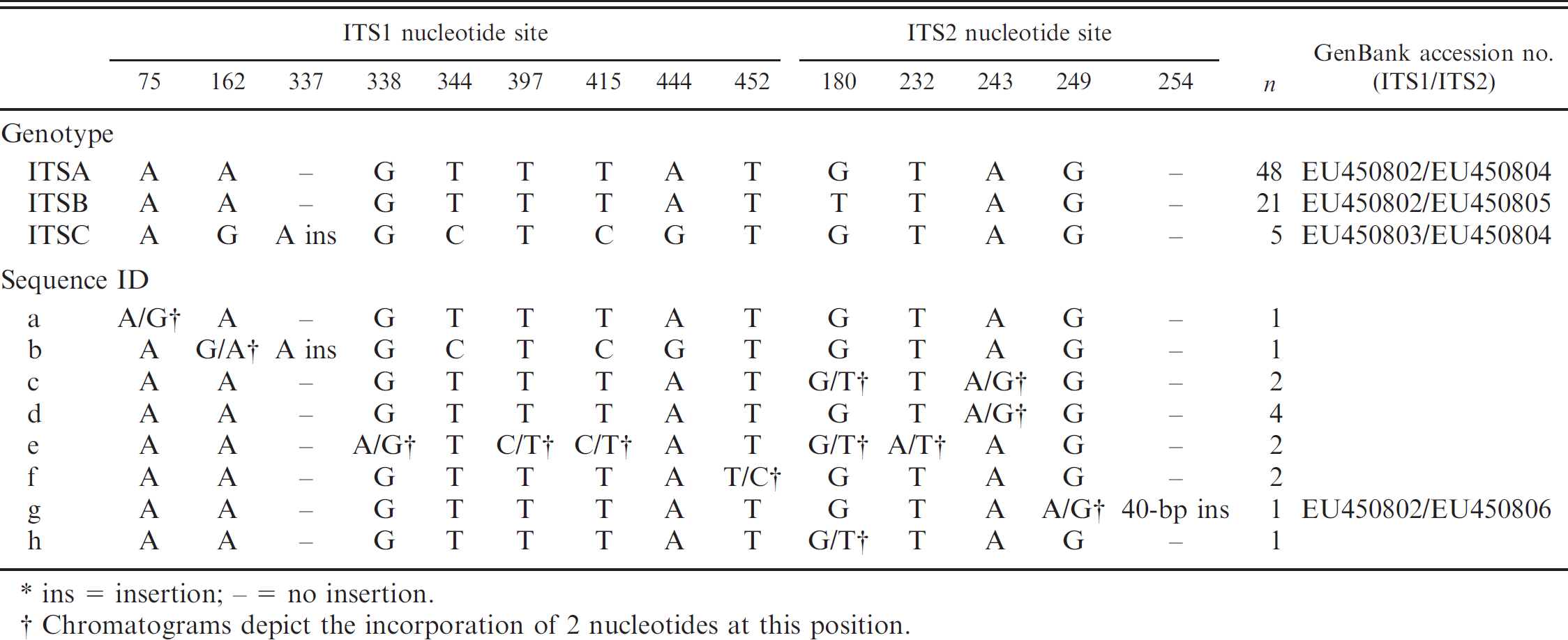
ins = insertion; – = no insertion.
Chromatograms depict the incorporation of 2 nucleotides at this position.
Results
PCR amplification and DNA sequencing
Amplification of the C. felis ITS regions from infected domestic cat blood samples and resolution of amplicons on agarose gel resulted in single bands of PCR product for 108 of 112 samples (93.4%). Sequencing of all products yielded unambiguous ITS1 and ITS2 sequence data for 88 of 108 samples (81.5%). Within the 458-bp ITS1 region of the C. felis genome, there were a total of 8 single nucleotide polymorphisms (SNP) and a single nucleotide insertion. Within the 265-bp ITS2 region of the C. felis genome, there were a total of 4 SNPs and a single 40-bp insertion. Taken together, the ITS1 and ITS2 sequence data define a total of 11 different sequences and 3 unique genotypes (Table 1). The most commonly identified ITS1-ITS2 genotype, designated as ITSA (GenBank accession nos. EU450802 and EU450804) was identified in 48 of 88 samples (54.5%). The second most common genotype, designated as ITSB (GenBank accession nos. EU450802 and EU450805) and differing by 1 SNP within ITS2, was detected in 21 of 88 samples (23.9%). The third genotype, designated as genotype ITSC (GenBank accession nos. EU450803 and EU450804) and differing by 4 SNPs and a single nucleotide insertion, was present in 5 of 88 samples (5.7%). The remaining ITS1-ITS2 sequences varied by 1–5 SNPs, in which the chromatograms revealed the incorporation of 2 nucleotides at a single position, and 1 sequence contained a 40-bp insertion within ITS2 (GenBank accession nos. EU450802 and EU450806). All samples with variable and mixed nucleotide SNPs and the 40-bp insertion were confirmed by repeated amplification and sequencing reactions.
Geographic distribution of genotypes
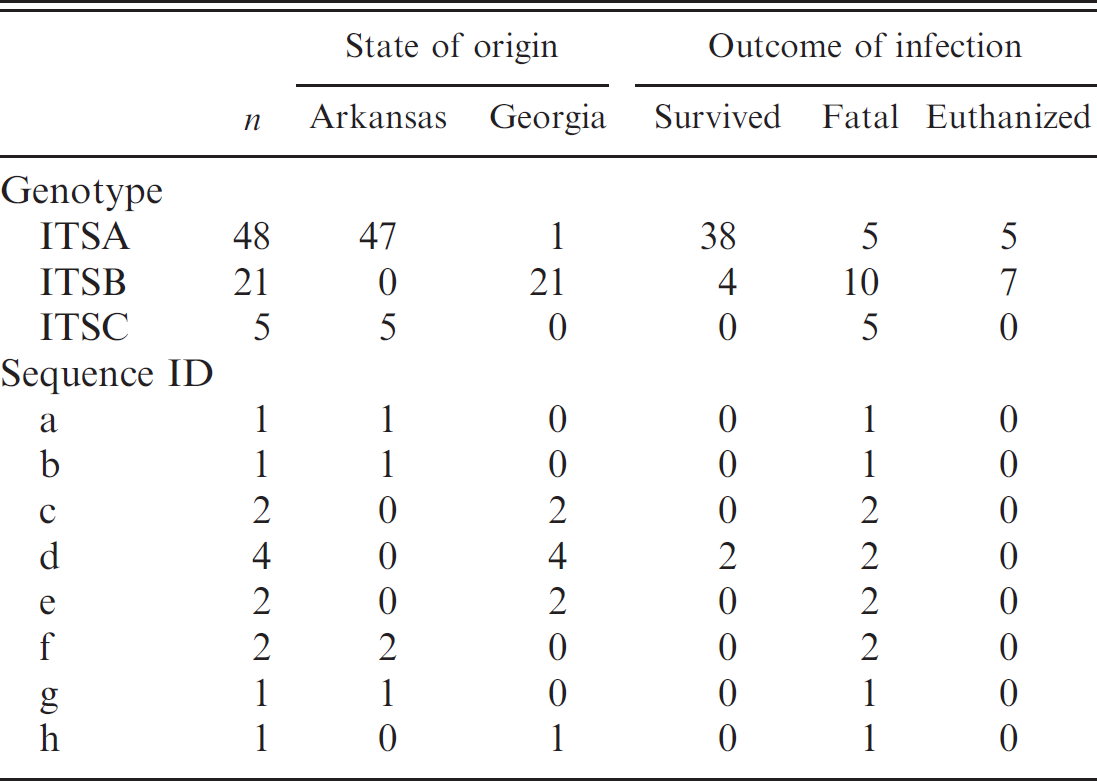
Overall, 57 of 88 sequenced C. felis samples (64.8%) were submitted from Arkansas, and 31 of 88 samples (35.2%) originated from Georgia. Among the unique ITS1-ITS2 genotypes, ITSA was detected in 48 of 57 Arkansas samples (84.2%) and 1 of 31 Georgia samples (3.2%). ITSB was detected in 21 of 31 Georgia samples (67.7%) and was not detected in any Arkansas samples, and ITSC was detected in 5 of 57 Arkansas samples (8.8%) and was not present in any Georgia samples. The geographic distribution of the ITS sequences is summarized in Table 2.
. State of origin and outcome of infection for Cytauxzoon felis first and second ribosomal internal transcribed spacer region (ITS1 and ITS2) genotypes and sequences.
Association of genotype with pathogenicity
Of the 88 C. felis samples analyzed, 12 were obtained from cats that were euthanized and were excluded from the analysis of clinical outcome of infection. Among those remaining, 44 of 76 samples (57.9%) were obtained from cats that survived infection. Among the unique C. felis ITS1-ITS2 genotypes, 38 of 48 (79.2%) of those infections identified as having the ITSA genotype survived compared with 4 of 21 (19.0%) of those infected with the ITSB genotype, and none of the 5 identified as infected with the ITSC genotype (P < 0.001). The clinical outcome for C. felis–infected cats with different ITS sequences is summarized in Table 2.
Discussion
Although cytauxzoonosis has historically been nearly 100% fatal in domestic cats, increasing reports of cats that demonstrated less-severe disease have led to the hypothesis that different strains of C. felis exist that vary in pathogenicity to the domestic cat. The detection of genetic variability within C. felis in the current study supports this hypothesis and reinforces the usefulness of mutation scanning to detect C. felis population variation. The level of nucleotide polymorphism detected in the current study is comparable with that detected among different isolates of Babesia canis species, a parasite closely related to C. felis. A 1998 study 18 that analyzed the sequence variability of the ITS1–5.8S-ITS2 rDNA regions among isolates of Babesia canis canis, Babesia canis vogelii, and Babesia canis rossi detected intraspecies nucleotide polymorphism on the level of 0.1–2.1%, which is similar to findings in the current study. This is a much smaller variability than that found between the different Babesia spp. (18–31 %) in the 1998 study, which supported the classification of the different genotypes detected in the current study as strains rather than separate species of Cytauxzoon.
Sequencing results of C. felis in the present study revealed unique ITS1-ITS2 genotypes that varied geographically. In particular, the ITSB genotype was detected only in samples submitted from Georgia and the ITSC genotype was detected only in samples from Arkansas. The identification of unique genotypes from distinct geographic areas might be expected in the development of different parasite strains. Although clean sequencing data were obtained for the majority of the C. felis ITS1 and ITS2 amplicons, samples in which either the ITS1 or ITS2 sequence was ambiguous were excluded from further analysis. A detailed review of the accepted sequencing data identified 14 samples for which the chromatograms revealed the incorporation of 2 nucleotides at a single position within ITS1 or ITS2, although this position varied among samples. This finding is interpreted to reflect coinfection of the cat with multiple C. felis genotypes. Alternatively, it is possible that the C. felis genome contains multiple copies of the rRNA genes with polymorphic units, as has been described for some isolates of the closely related parasite, Theileria parva. 10 However, in the current study, separate C. felis samples that reflect a single infection with each of the 2 different alleles presumed to combine in the mixed infection were detected for some of the mixed nucleotide SNPs. Although not detected for all SNPs, this inconsistency may be a consequence of the study's sample size, and further analysis of more C. felis genotypes from these geographic areas may detect infection with each unique parasite allele. Because of the inability to discern between each genotype's influence on clinical outcome in the cats with suspected coinfection with multiple parasites, the sequences that contained the nested double peaks in the present study were excluded from the analysis of the association of genotype and pathogenicity.
A strong association was detected between particular C. felis ITS1–ITS2 genotypes and survival of domestic cats after acute infection. The C. felis ITSA genotype is of special interest, given its association with a very high survival rate, particularly when compared with the high fatality rate for infection with other C. felis genotypes. Thus, ITS sequencing may be useful to define markers that are able to detect more or less pathogenic strains of C. felis among infected cats. Interpretation of the association of C. felis genotype and clinical outcome in infected domestic cats, however, is limited in the present study, which does not account for other factors that may affect clinical outcome. For these clinical cases, there was no control for the differences in treatment regimens, and the study design does not account for differences in host factors, such as host genetics and immune response, that may play a significant role in pathogenicity of cytauxzoonosis in infected cats. Still, a statistically significant association of certain ITS genotypes with clinical outcome of infection has been documented.
At present, only the ribosomal genes have been identified and sequenced for C. felis, which limit the areas of the genome that can be investigated for sequence variability, and the design of the current study precludes identifying genetic variability outside of the ITS regions. Ultimately, identification of genetic variability in additional loci, ideally in potential pathogenicity-determining genes, would best test the hypothesis that different C. felis strains exist that vary in pathogenicity to the domestic cat. Although identification of virulence-associated genes is not a simple task, it should be feasible to use genomic data from related piroplasms, such as Babesia bovis and T. parva, to identify additional loci to be characterized with the assumption that one or more new loci may be more closely linked to potential virulence genes. Further genomic studies of C. felis are currently limited by the inability to isolate parasite DNA free from the infected host DNA.
The current study revealed sequence variability in the ITS1 and ITS2 regions of C. felis from clinically ill domestic cats diagnosed with cytauxzoonosis. Unique ITS genotypes were identified that varied geographically between Arkansas and Georgia. In addition, there was a strong association between particular ITS genotypes and the outcome of infection. The findings support the hypothesis that there are genetically distinct populations of C. felis, which may account for the variability in the clinical outcome of domestic cat infections. A better understanding of the epidemiology of C. felis infection will enhance the ability to prevent or treat this highly fatal infectious disease.
Acknowledgements
The authors wish to thank Mike Mahoney and Mariann Clark (Arkansas Livestock and Poultry Commission Laboratory, Little Rock, AR) and Dr. Debra Miller and Anita Merrill (Veterinary Diagnostic and Investigational Laboratory, UGA, Tifton, GA) for their efforts in providing samples of C. felis to be used in this study. Funding for this project was provided by The Morris Animal Foundation and the UGA Clinical Research Program.
Footnotes
a.
Illustra blood genomicPrep Mini Spin Kit, GE Healthcare, Little Chalfont, Buckinghamshire, UK.
b.
HotStarTaq Plus Master Mix, Qiagen Inc., Valencia, CA.
c.
QIAquick PCR Purification Kit, Qiagen Inc., Valencia, CA.
d.
MWG Biotech Inc., High Point, NC.
e.
University of Georgia Integrated Biotechnology Laboratories, Athens, GA.
f.
Invitrogen Corp., Carlsbad, CA.