
Abstract
The BVDV envelope glycoprotein Erns/gp48 and the C terminal 79 amino acids of the capsid protein coding region were expressed in a baculovirus system and antigenically characterized. Western blot assay was used to detect recombinant Erns (r-Erns) in infected insect cells using specific monoclonal antibodies. The r-Erns was then used in an indirect ELISA to detect BVDV specific antibodies in a panel of 540 well-characterized sera. Results of the r-Erns ELISA were compared to those obtained with a commercially available competitive ELISA targeting anti-NS2/3 antibodies. A good correlation was observed between the 2 ELISA (kappa = 0.916, 95% C.I.: 0.876, 0.956). Using the commercial NS2/3 ELISA as the reference test, the relative sensitivity of r-Erns ELISA was 97.5% (95% C.I.: 94.3%, 99.1%) and the relative specificity was 93.9% (95% C.I.: 89.4%, 96.9%), while relative specificity was 100% (95% C.I.: 97%, 100%) using true negative sera (derived from a negative herd). All but 1 antigen positive animals (n = 36) tested negative in the r-Erns ELISA; among them all 22 confirmed PI animals were negative by r-Erns ELISA. The ability of r-Erns ELISA to identify cattle immunized with inactivated vaccine was also demonstrated in a small group of cattle, compared to an NS2/3 antibody ELISA. Results suggest that r-Erns ELISA represents an alternative test for antibody generated by natural infection or BVDV vaccination.
Introduction
Bovine viral diarrhea virus (BVDV) is classified in the pestivirus genus of the Flaviviridae family. Members of this genus include classical swine fever virus (CSFV) and border disease virus (BDV). 11 BVDV is responsible for reproductive, enteric, and respiratory syndromes and causes considerable economic loss to cattle farms throughout the world. 8,26,27 The infection is sustained in the population by persistently infected (PI) animals 14,18 that are immunotolerant to the persisting virus strain due to initiation of infection early in their intrauterine development. 1,8 This unique phenomenon is the primary mechanism whereby BVDV is maintained in cattle populations, providing for direct and indirect transmission. 6 Such PI animals shed the virus through secretions and excretions and may, over months or years, develop an invariably fatal disease known as mucosal disease as a result of mutation of persisting virus or superinfection with an antigenically related heterologous strain. 7 Therefore, control and eradication programs must focus on prevention of persistent infection and identification and removal of PI animals. 5 Although PI animals constitute a major source of virus spread, transiently infected animals may be the primary source of virus introduction into naive herds. In most cases, the transient infection produces reproductive and respiratory disease, which results in major economic losses to the cattle industry. However, occasionally severe disease characterized by hemorrhages is observed. 13,24,27,30,31,34
In low cattle population density areas, eradication of BVDV without vaccination has proven to be successful, whereas in high population density or high seroprevalence areas, control measures are usually carried out by systematic vaccination of cattle. 17 It should be borne in mind that, due to antigenic variability, vaccination alone cannot guarantee full protection against all viral strains, and there is need to improve the efficacy and safety of BVDV vaccines. 16,35
A number of diagnostic tests have been developed to detect antibodies or viral antigens or nucleic acid at the herd and individual level. Infected herds are usually identified by spot test (testing animals of a certain age or groups of animals housed in different units), bulk milk test for antibodies, or nucleic acid amplification. 17,25,28 Once the infected herds are identified, individual tests are used to remove PI animals. In regard to the presence of circulating antibodies or virus, 3 main categories of animals can be differentiated: 1) seropositive and virus negative animals due to previous transient infections or vaccination, 2) immunologically naive animals, and 3) virus-positive animals as a result of persistent or transient infection. 6 PI animals usually remain negative for antibody against the conserved epitopes of structural and nonstructural proteins; however, a superinfection with an antigenically different BVDV strain may induce an immune response against variable epitopes, mainly located in the E2 envelope glycoprotein. 3,16 Serological tests are available to detect antibodies against both variable proteins (serum neutralization test (SNT)) and conserved proteins (competitive NS2/3 ELISA). The latter test, based on a monoclonal antibody directed against a highly conserved epitope of NS2/3, has proven to be highly sensitive in detecting antibodies in transiently infected animals or animals vaccinated with a live attenuated vaccine. 2 However, due to insufficient amount of this nonstructural protein in most killed vaccines, this test may fail to detect antibodies in animals that have been immunized with inactivated vaccine.
Among viral structural proteins the Erns glycoprotein has been shown to be a useful marker for identification of PI animals, since it is genetically and antigenically conserved among different isolates. 22 Indeed, several studies have indicated that some of the immunologically relevant Erns epitopes are conserved among different BVDV isolates, 4,12,23,36 and few differences are found in Erns amino acid composition among different pestiviruses. 19 Little is known about its potential as a serological marker of BVDV infection. Recombinant Erns or its subunit, expressed in prokaryotic and baculovirus systems, has been used as an antigen to specifically detect anti Erns antibodies in a limited number of samples with promising results, 23,29,37 but the application on a large number of well-characterized samples is still necessary for more complete validation. In this study, the Erns and the C terminal 79 amino acids of the capsid protein coding region were expressed in a baculovirus system. The resulting antigen was used to develop an indirect ELISA that was validated using a panel of sera from transiently infected animals, animals immunized with inactivated vaccine, PI animals, and immunologically naive animals.
Materials and methods
Virus and cells
The Oregon C24 strain of BVDV was cultured in Madin Darby bovine kidney (MDBK) cells maintained in Earle minimal essential medium supplemented with 10% pestivirus-free fetal calf serum a and antibiotic-antimycotic solution (penicillin 100 Ul/ml, streptomycin 100 μg/ml, amphotericin B 0.25 mg/ml). Linearized Autographa californica multiple nuclear polyhedrosis virus (AcMNPV) DNA was used to generate recombinant baculovirus. Spodoptera frugiperda (Sf9 or Sf21) cell lines a were grown in TNM-FH Insect Medium (Modified Grace Medium) k with 10% fetal calf serum, antibiotic-antimycotic solution, and were used for transfection and plaque purification and to generate high-titer recombinant viral stocks. The SF21 cell line was adapted to Sf900II protein-free medium according to a direct method protocol a and used for time-course expression and large-scale expression of recombinant protein.
Construction of recombinant baculoviruses
Total RNA was extracted from infected MDBK cell cultures using RNeasy total RNA kit b and reverse transcribed in a 20-μl reaction containing 20 U AMV reverse transcriptase, c 0.1 A260 U random hexamers, 1 mM dNTPs, and 2–3 μg total RNA, according to the manufacturer's protocol. The primers used to amplify the Erns gene and part of the capsid protein coding region were: forward primer 5′ TTGGATCCATGAAAATAGTGCC-CAAAGAGT 3′, reverse primer 5′ TTGAATTCAGCCG-CATATGCTCCAAACCACGT 3′. Primers contained the BamH1 and EcoR1 recognition site (underlined), respectively, at the 5′ end to facilitate cloning. Five ml of cDNA was added to 45 μl of PCR mix containing 10 mM Tris-HCl, 50 mM KCl, 2 mM MgCl2, 0.2 mM dNTPs, 10 pM of each primers, and 2 U of AmpliTaq Gold. d Thermocycling parameters consisted of an initial denaturation step (94°C, 10 min) followed by 40 cycles of denaturation (94°C, 1 min), annealing (50°C, 1 min), and extension (72°C, min). The purified PCR product was digested with BamHI and EcoRI restriction enzymes and ligated into the baculovirus shuttle vectors pBlueBacHis2A and pMel-BacA. a The former allows purification of protein of interest using metal chelate affinity chromatography, while the latter directs the expression of the recombinant protein through the secretory pathway to the extracellular medium. Escherichia coli TOP10 a was transformed with ligation mixtures and used to generate midipreparation (100-mg scale) of each recombinant plasmid. Sequence analysis of the insert was carried out with fluorescent dye terminators on an ABI PRISM 310 Genetic Analyzer d according to the manufacturer's protocol. Purified plasmids were used to cotransfect Sf9 insect cells with linearized AcMNPV DNA using liposome reagent cellfectin. a After 3 days of incubation at 27°C, the supernatant was harvested and recombinant virus was purified by plaque assay. Positive plaques were selected and amplified in a 24-well microplate. After 7–10 days, a PCR control was carried out using vector primers designed to bind the flanking region of the insert, allowing the detection of recombinant baculovirus (expected length 1,259 bp) as well as any contaminating wild-type baculovirus (expected length 839 bp). Recombinant baculovirus was used to infect Sf21 insect cells to generate high-titer viral stock, which was titrated by plaque assay. Recombinant baculoviruses obtained using pBlue-Bac and pMelBac vectors were identified as Bac/Erns and Mel/Erns, respectively. All technical methods were carried out according to the manufacturer's instruction manual. a Location and peak of expression were estimated using a sandwich ELISA, e which was developed for identification of PI animals through specific detection of native Erns in serum samples.
Production and characterization of recombinant antigen
Sf21 cells were infected with the recombinant baculovirus at a multiplicity of infection (MOI) of 10 PFU/cell. Cells were harvested 72 hr postinfection, washed with phosphate-buffered saline (PBS), and used as whole antigen for Western blot analysis, using commercially available anti-6xHis f and anti-Erns e monoclonal antibodies (MAbs). Western blot was carried out following conventional methods. 9 For antigen preparation, Sf21 cells were infected with the recombinant baculovirus at a multiplicity of infection (MOI) of 10 PFU/cell. At 72 hr postinfection, cell monolayers were washed with PBS and lysed with 150 mM NaCl, 50 mM Tris HCl pH 8.0, and 1% Igepal. The mixture was first incubated at −80°C for 1 hr and, after thawing, clarified at low-speed centrifugation. Supernatant was further centrifuged at 8000 × g for 20 min at 4°C and stored in aliquots at −80°C. The protein concentration of the total cell lysate was estimated by DC Protein Assay. g
Serum samples
A panel of 540 bovine sera, collected mainly from 6 different Italian herds, was used in this study. Among these, 100 serum samples were collected from true negative herds. 24,26 The negative status of these herds was determined using SNT and NS2/3 competitive ELISA, by testing either all animals or a sample size that allowed the detection of prevalence of infection ≥10% (95% confidence level). 10 Thirty-six sera were from antigen-positive subjects, identified from different herds in Northern Italy by antigen ELISA or RT-PCR, and 22 of those were proven to be PI animals by positive retesting after 2 weeks; 384 sera were from infected farms where at least 1 PI animal had been detected. Among the latter group seropositives and seronegatives were previously classified using a commercially available competitive ELISA detecting anti-NS2/3 antibodies. i Finally, 20 sera were collected from 10 animals, aged 6–8 mo, immunized with 2 doses of inactivated vaccine h at a 1-mo interval. Sera were collected before vaccination and 1 mo after the second administration.
Recombinant Erns protein-based antibody ELISA
For r-Erns antibody ELISA, 2 different coating procedures were tested. The first procedure consisted of coating microplates overnight at 4°C with 1 mg/well (odd columns) of infected cell lysate diluted in carbonate/bicarbonate buffer, or the same amount of negative antigen (even columns), prepared from mock infected Sf21 cells. After 3 washes with ELISA wash (0.125 mM NaCl, 0.05% Triton × 100) microplates were used for ELISA. In the second procedure, microplates were coated overnight at 4°C with 1 mg/well of an anti-Erns noncompeting Mab j diluted in carbonate/bicarbonate buffer. The next day, plates were washed twice and blocked with casein buffer (2.5% casein, 0.375 mM NaCl pH 7) at 37°C for 1 hr. Following four washing steps, the insect cell lysate containing expressed Erns was diluted to 2 μg/well in PBS and added to MAb-coated plates. After 1 hr incubation at 37°C, plates were washed 3 times and used for ELISA. In a preliminary phase both coating methods were used to analyze a subset of serum samples, then the second coating method was chosen. Duplicate antigen-coated wells were incubated for 1 hr at 37°C with test serum diluted 1/40 in PBS containing 1.25% casein. Plates were washed 3 times and incubated with 5 ng/well of peroxidase-conjugated anti-bovine IgG (diluted in the same buffer as before) for 1 hr at 37°C. After the final washing step, reaction was developed with 2,2′-azino-bis(3-ethylbenziazoline-6-sulfonic acid) (ABTS) for 20 min and optical density (OD) measured at 405 nm. The mean OD from duplicate wells of each sample was expressed as a percentage of the OD of the positive control. The latter was a pool of 4 serum samples positive in the NS2/3 ELISA.
Statistical analysis
Receiver operating characteristic (ROC) analysis of the ELISA results was performed using the R software. 20 An optimum cutoff, corresponding with the highest accuracy (i.e., minimal false-negative and false-positive results), was determined. Serum samples yielding ≥ 25% of the OD of the positive control (included in each plate) were considered to be positive. Concordance between r-Erns ELISA and the commercial NS2/3 competitive ELISA was evaluated using the Kappa statistic, 15 while descriptive statistics and distribution of reactivity of each group of samples were obtained using the R software. 20 Relative sensitivity and specificity were calculated using binomial test function in R. 20
Results
Results demonstrated that Bac/Erns and Mel/Erns infected cells were both highly reactive by sandwich ELISA, with an expression peak at 72 hr p.i. (post infection) and at 96 hr p.i., respectively (Fig. 1). However, in Bac/Erns infected cells, the signals were mainly cell-associated, whereas in Mel/Erns infected cells, r-Erns was found secreted in the medium. Western blot analysis of total cell lysate, using anti-Erns and anti-6xHis MAbs, revealed a major protein band with a molecular mass slightly lower than the expected size (Fig. 2). Preliminary attempts to obtain r-Erns in purified form from Bac/Erns infected cells using affinity chromatography under native or denaturing conditions were unsuccessful. Recombinant Erns, obtained in secreted form into the SF900II medium using Mel/Erns infected cells, was found to rapidly degrade after storage at −80°C. Therefore, antigenic characterization of r-Erns as a serological marker of BVDV infection was carried out using a crude Bac/Erns infected cell lysate. When crude cell lysate was used for direct coating of microplates, a number of false positive reactions were recorded in spite of correction for background staining by subtracting the OD of negative antigen. Using a noncompeting MAb as a capture antibody, no false positives were detected using true negative samples; therefore, the characterization of the entire serum panel was carried out using that method. Distribution of ELISA ODs for each group of serum samples is shown in Fig. 3. The specificity of Erns ELISA using 100 true negative sera was 100% (95% C.I.: 97%, 100%). Among 36 antigen positive animals, all were negative by NS2/3 ELISA; samples from all 22 PI animals and 13 out of 14 antigen positive samples (not proven to be PI animals) were also negative by Erns ELISA. The relative specificity and sensitivity of Erns ELISA, using 182 negative sera and 202 positive sera previously classified by NS2/3 ELISA, were respectively 93.9% (95% C.I.: 89.4%, 96.9%) and 97.5% (95% C.I.: 94.3%, 99.1%). Agreement between Erns ELISA and NS2/3 competitive ELISA, evaluated on 3 infected farms, was excellent (kappa = 0.916, 95% C.I.: 0.876, 0.956). When the ability of Erns ELISA to detect animals immunized with inactivated vaccine was evaluated 1 month after the booster vaccination, all samples scored negative to NS2/3 ELISA, while 9 out of 10 showed a clear seroconversion against r-Erns (Fig. 4).
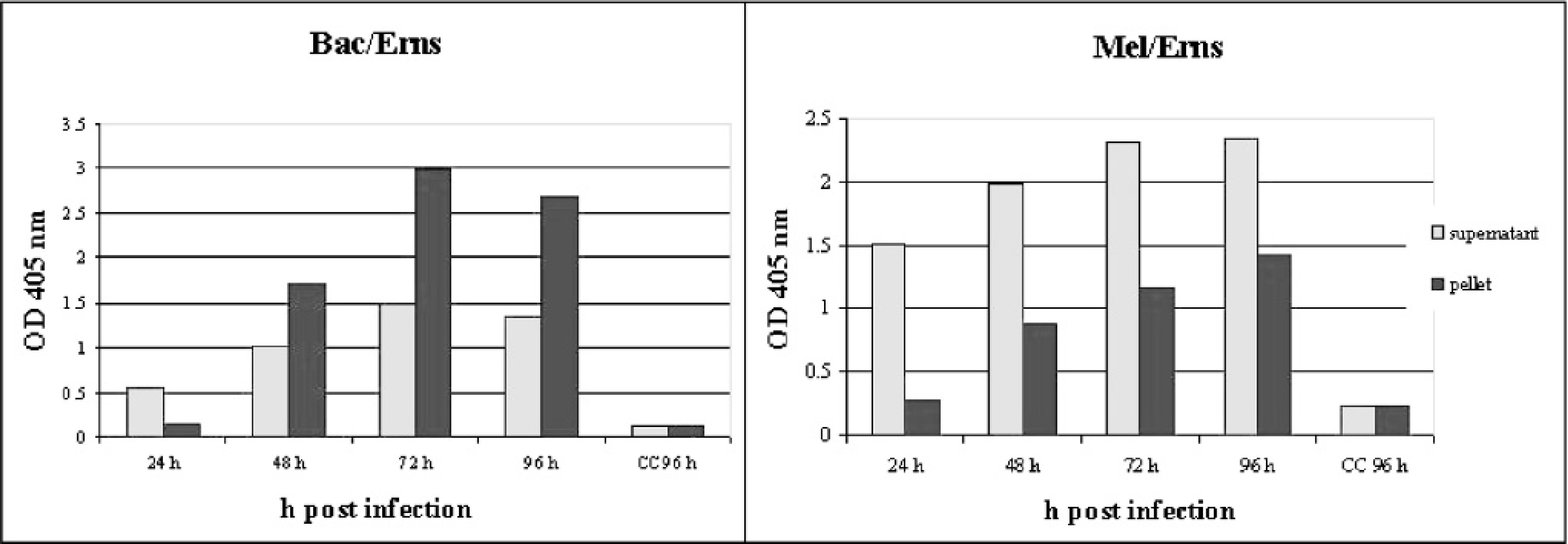
Time course expression and localization of r-Erns in Bac/Erns and Mel/ Erns infected insect cells, revealed by sandwich ELISA. e
Discussion
In this study, the diagnostic value of r-Erns expressed in baculovirus as a serological marker of BVDV infection was evaluated. The rationale for this approach was based on 2 properties of the Erns. First, the gene encoding this glycoprotein is relatively conserved in the different viral strains of both genotypes. 19,23,32 Second, Erns is able to induce a rapid immune response during viral infection in immunocompetent animals. 23 The baculovirus expression system is known to generate a high amount of viral glycoprotein preserving major posttranslational modifications and antigenicity. 21 Immunotolerant and persistently infected animals should remain antibody negative against conserved epitopes and should be nonreactive against this glycoprotein. Several studies demonstrate that PI animals are able to respond to BVDV immunogens different from the specific BVDV inducing immunotolerance. 3,16 However, in these studies the presence of antibodies in PI animals was exclusively detected using the SNT when a PI subject is superinfected with an antigenically different BVDV strain. Although Erns may induce neutralizing antibodies, E2 is the most important structural protein involved in the production of such antibodies. This glycoprotein, unlike Erns, is highly variable among genotypes and, to some extent, within subtypes. 16 Changes in the E2 glycoprotein are the primary sites of variation in neutralizing epitopes. 6
In the system used in this study, the r-Erns was efficiently expressed and the infection of a 100-mm monolayer plate (about 12 × 10 6 cells) produced enough antigen to test 4,400 sera using the described conditions. Preliminary antigenic characterization using 2 anti-Erns MAbs showed that at least 2 epitopes were preserved in the recombinant protein. Native Erns lacks a typical membrane anchor and is released in a soluble form in viremic subjects. 32,33 This property was also observed in the r-Erns and further enhanced using Mel/Erns infected cells in which the upstream signal sequence for honeybee mellitin is used to direct secretion of the recombinant protein.
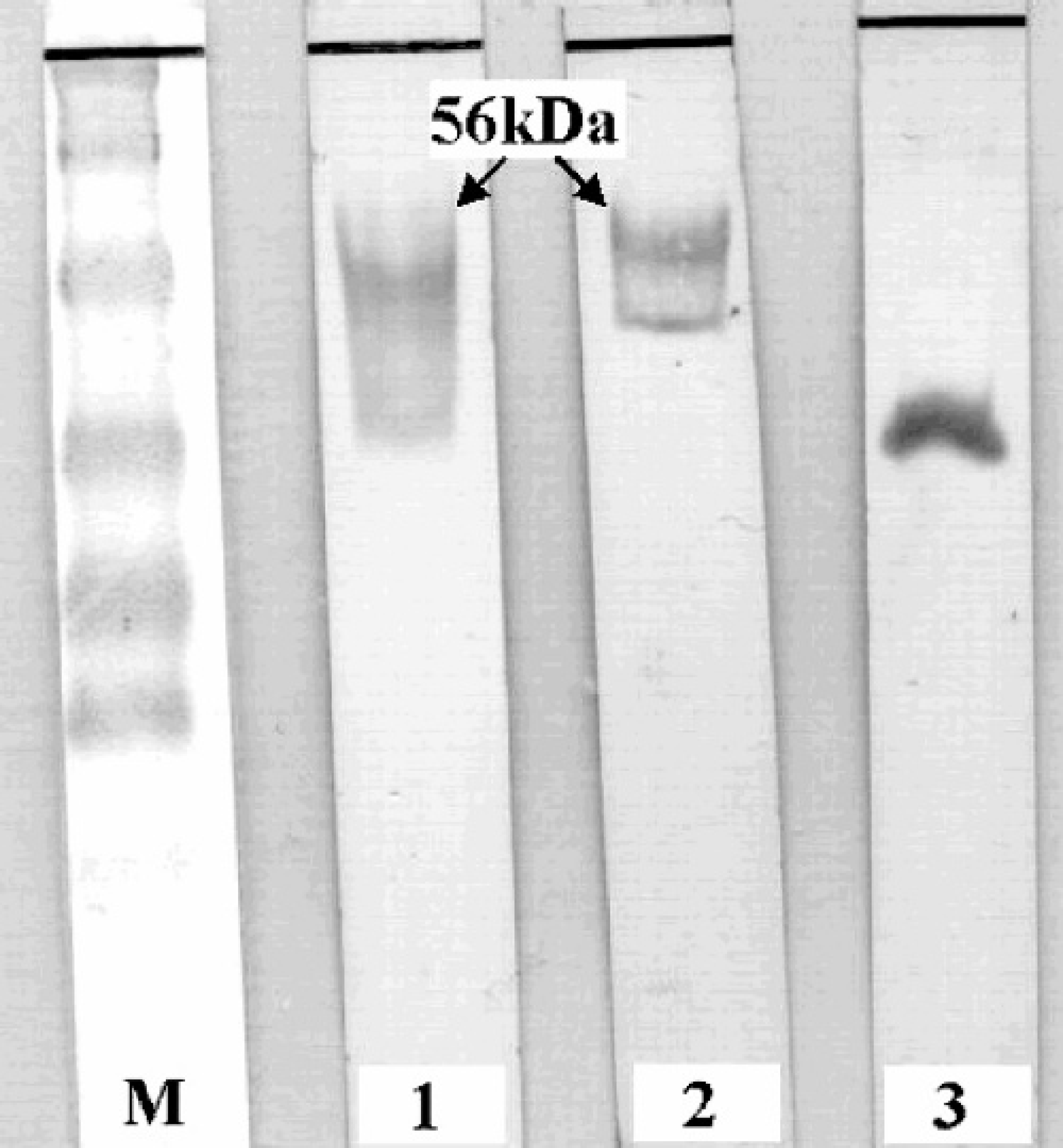
Western blot analysis of Bac/Erns infected insect cells at 72 hr p.i. M: prestained MW markers; lane 1: anti-6xHis MAb; lane 2: anti-Erns MAb; lane 3: 6xHis control recombinant protein.
The use of crude insect cell lysate has been suggested as a practical advantage for the production of diagnostically useful antigen for BVDV diagnosis due to the low level of nonspecific reactions against insect proteins. 29 However, the results from this study indicate that purification of the recombinant antigen from crude insect cell lysate might be required for optimal specificity in an indirect ELISA system. However, the need to purify r-Erns was obviated by utilizing a commercially available MAb to capture BVDV-specific antigen from the crude cell lysate. R-Erns ELISA showed 100% specificity when used to test 100 negative samples recovered from farms where the negative BVDV status had been clearly determined. All samples from 22 PI animals, supposed to be antibody negative against conserved epitopes, were also negative in the Erns ELISA. A lower value of specificity was obtained in comparison with NS2/3 ELISA (93.9%). While attenuated vaccine may induce an antibody response against structural and non-structural viral protein similar to transient infection, inactivated vaccine may induce response only against structural proteins. This may reflect the ability of Erns ELISA to identify animals immunized with inactivated vaccine, a common control measure in Italian farms, and confirms a previous study carried out on recombinant BVDV antigen expressed in prokaryotic and baculovirus systems. 23 To further confirm this hypothesis, a group of animals was vaccinated with 2 doses of inactivated vaccine, and a clear seroconversion was recorded by r-Erns ELISA but not by NS2/3 ELISA. The inactivated vaccine used for immunization in this study was tested by a sandwich ELISA to detect the presence of native Erns and a very high signal was detected (data not shown), suggesting that Erns was highly expressed and preserved during vaccine preparation. The low immunogenic properties of NS2/3 in killed vaccines may be related to trace amounts of this nonstructural protein in vaccine preparation due to a lower expression, compared to structural proteins, and/or removal by final filtration in some vaccine preparations. The fact that Erns lacks an anchor site may lead one to assume an NS2/3-like behavior. It will therefore be necessary to evaluate different killed vaccine preparations to confirm the ability of the test proposed in this study to identify immunized animals. When the sensitivity of r-Erns ELISA was evaluated using sera from infected herds and previously classified by NS2/3 ELISA, excellent agreement was observed. Finally, all but 1 viruspositive animal did not react against r-Erns ELISA. The lone reactor was a 2-year-old cow belonging to an infected herd which was slaughtered before it could be retested to ascertain PI status. If this animal was transiently infected, it is possible that it might have been tested during a very narrow window in which viral antigen (captured by the noncompeting MAb) and anti-Erns antibodies were present at the same time. However, since neutralization test and NS2/3 antibody ELISA were both negative, a false-positive reaction with the r-Erns ELISA likely occurred in a true PI animal or in an animal during the early stages of a transient infection. With the exception of this possible false positive, the r-Erns ELISA proved to be a highly reliable antibody test.
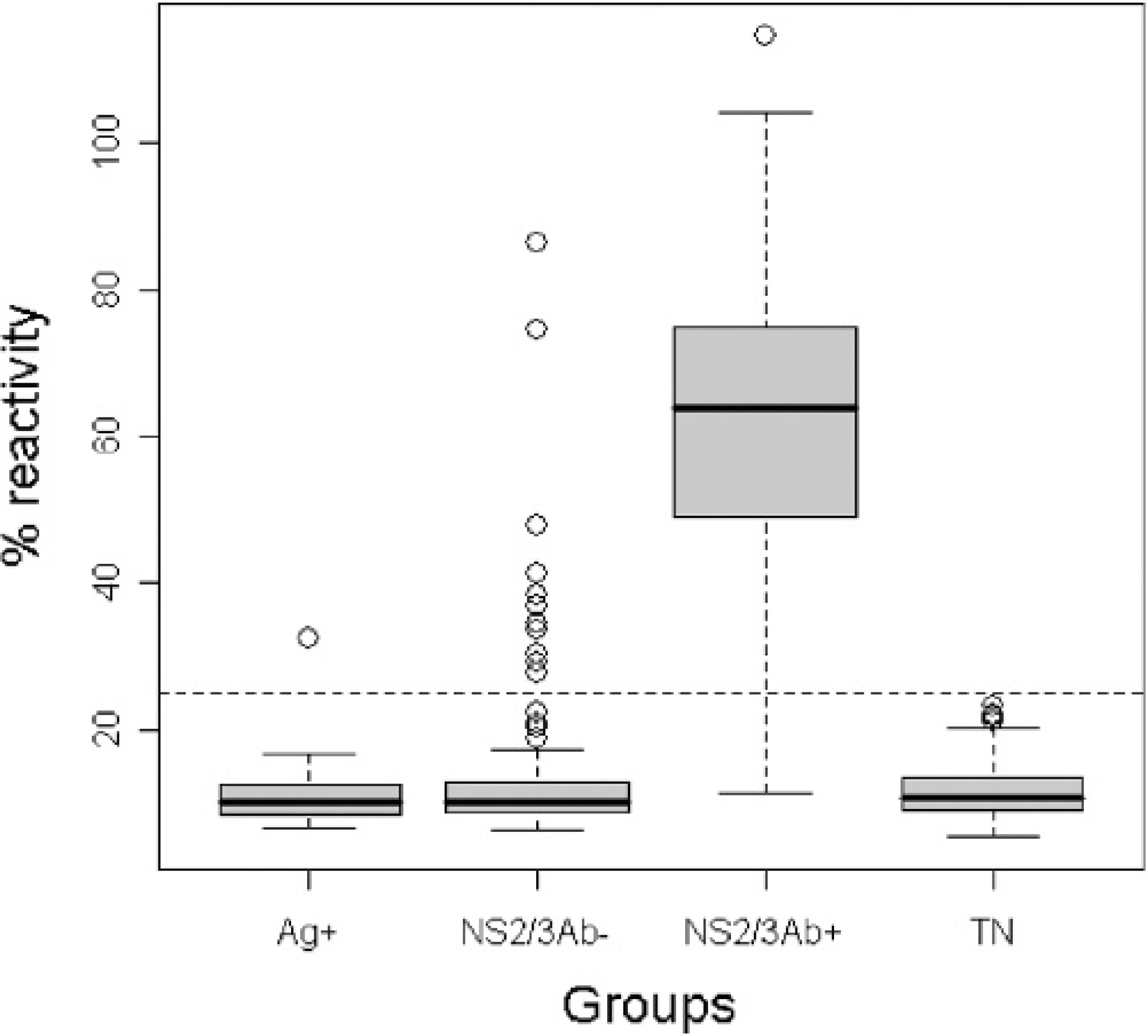
r-Erns ELISA. boxplot of mean ODs (expressed as percentage of reactivity of the positive reference serum) of the serum panel used in the study; Ag+: 36 virus positive animals (by antigen or nucleic acid detection) of which 22 were PI animals; NS2/3Ab- and NS2/3Ab+: 384 sera from infected farms classified by NS2/3 ELISA; TN: 100 truly negative sera.
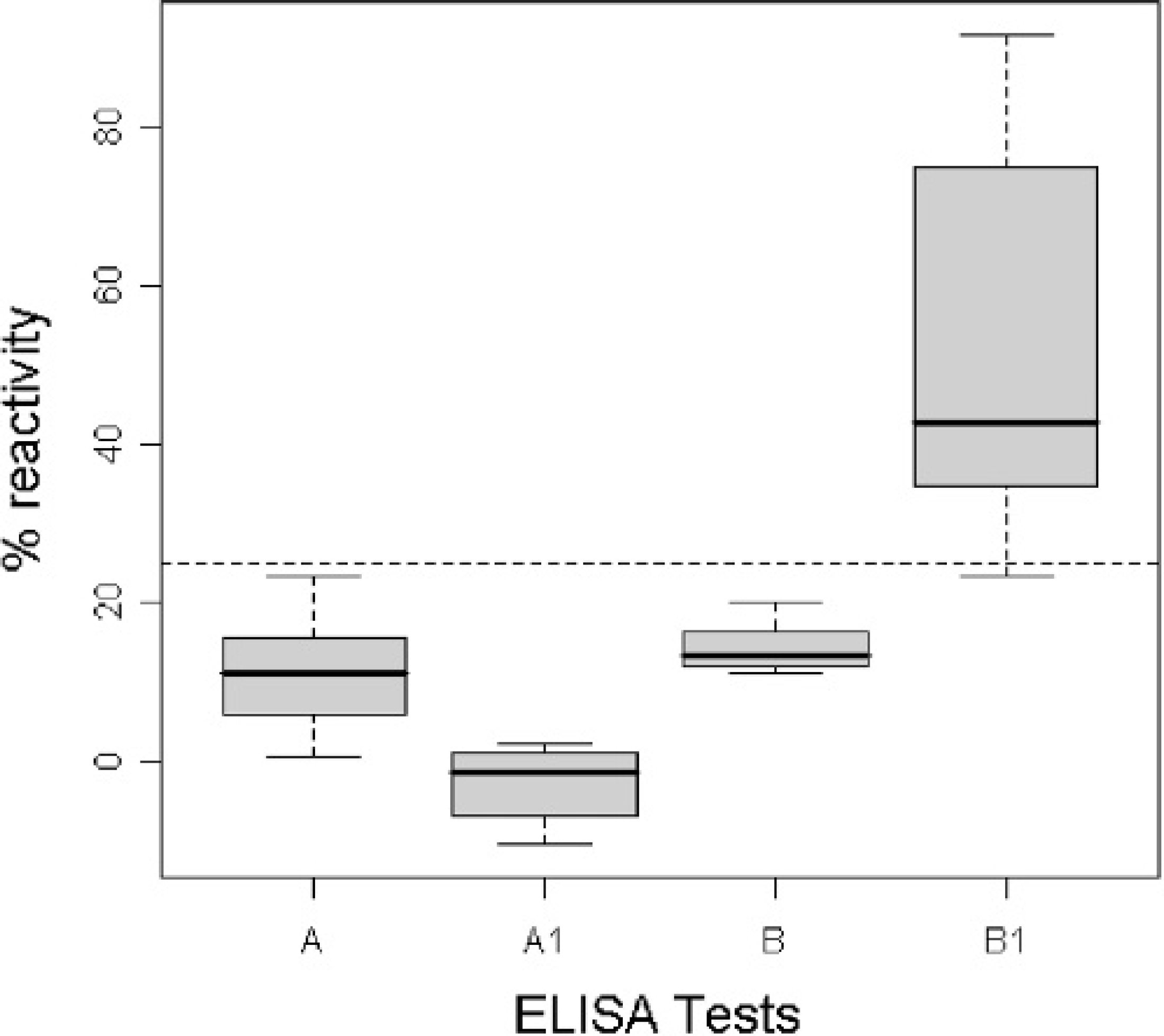
Boxplot of mean ODs (expressed as percentage of reactivity of the positive reference serum) of 20 sera collected from 10 cows. A and B represent 10 sera collected before vaccination and tested with NS2/3 ELISA and r-Erns ELISA respectively. A1 and B1 represent 10 sera collected after vaccination and analyzed with the same tests. Cutoff values were normalized for the 2 assays.
In conclusion, the Erns protein, expressed in a baculovirus-based eukaryotic system, preserves the antigenicity of the native protein. The ELISA described herein based on the baculovirus r-Erns protein may be a useful alternative serological test for detecting antibodies from transient infections or vaccination with BVDV.
Acknowledgements
We thank Ernst Peterhans for reviewing the manuscript. We also are grateful to Bommeli Diagnostics for providing anti-Erns monoclonal antibodies. This work was supported by Regione Piemonte, Progetti di Ricerca Sanitaria Finalizzata.
Footnotes
a.
Invitrogen, Carisbad, CA.
b.
Qiagen, Hilden, Germany.
c.
Finnzymes, Espoo, Finland.
d.
Applied Biosystems, Foster City, CA.
e.
Chekit BVD-VIRUS II, Bommeli Diagnostics, Bern, Switzerland.
f.
Amersham Biosciences, Little Chalfont, UK.
g.
Biorad, Hercules, CA.
h.
PregSure Pfizer Animal Health S.A, Louvain-la-Neuve, Belgium.
i.
Istitut Pourquier, Montpellier, France.
j.
Bommeli Diagnostics, Liebefeld-Bern, Switzerland.
k.
Sigma-Aldrich, St. Louis, MO.