
Abstract
Feces are increasingly valued as practical samples for molecular diagnosis of infectious disease. However, extraction of polymerase chain reaction (PCR) quality DNA from fecal samples can be challenging because of coextraction of PCR inhibitors. Because the type and quantity of PCR inhibitors is influenced by diet, endogenous flora, and concurrent disease, it is unlikely that extraction method performance with human feces can be directly extrapolated to that of domestic cats. In the present study, 4 commercially available DNA extraction methods were examined for their influence on the sensitivity of PCR for the detection of Tritrichomonas foetus in feline stool. DNA was extracted from serially diluted feline-origin T. foetus trophozoites in the absence or presence of feline feces. The ZR Fecal DNA kit was identified as affording the greatest analytical sensitivity and reproducibility and was able to detect ≥10 T. foetus organisms per 100 mg feces in 100% of PCR reactions. Further, the identified extraction method could be completed in the shortest time of all kits tested.
Feces are increasingly identified as practical samples for molecular diagnosis of infectious disease. 1,2,5–8 However, relative to other biological samples (e.g., blood) the composition of feces is highly complex. Accordingly, consistent extraction of high-quality DNA from fecal samples can be quite challenging because of the presence of polymerase chain reaction (PCR) inhibitors that are coextracted with DNA. These PCR inhibitors include complex polysaccharides, bile salts, hemoglobin degradation products, polyphenolic compounds, and heavy metals. Further, the type and quantity of PCR inhibitors present vary with composition of the feces, which is largely influenced by species, diet, and concurrent disease. Therefore, a functional PCR assay for pathogen detection in fecal samples must address these inherent issues during the DNA extraction process. In recent years, the use of commercial kits specifically designed to isolate DNA from fecal specimens have become increasingly popular. Each incorporates a method for removal of PCR inhibitors from the sample. Commercial kits vary in price and length of procedure, as well as in user-friendliness. In the present study, the efficacy of 4 commercially available DNA extraction kits a–d for the detection of Tritrichomonas foetus in feline stool specimens by PCR was compared. Each kit differed in the method used for removal of PCR inhibitors. For kit A, a extracted DNA is centrifuged through an inhibitor removal chromatography resin. For kit B, b the fecal lysate is incubated with an InhibitEX tablet that adsorbs inhibitors and is subsequently removed by centrifugation. For kit C, c an “inhibitor removal solution” is included during fecal sample lysis. For kit D, d a prewash solvent buffer and an inhibitor-retaining filtration step are used. For 3 of the 4 methods, studies that examined their efficacy for extraction of PCR-quality DNA from mammalian fecal specimens have not been previously reported.
To account for individual variation among cats, feces were obtained from three 1-year-old neutered males fed the same diet ad libitum. e For each cat, DNA was extracted in sextuplicate from 50-mg (kit A), 180-mg (kits B and C), 150-mg, and 100-mg (kit D) aliquots of feces. Before extraction, each fecal sample was spiked with 0, 1, 10, 100, 1000, or 10,000 feline T. foetus organisms contained in a 20-μl volume. Briefly, cultured T. foetus were washed in sterile saline solution, pelleted, and resuspended at a concentration established by counting a formalin-fixed aliquot of the organisms in a hemacytometer. All fecal samples were spiked with T. foetus derived from the same serial dilution series. Accuracy of the serial dilutions was confirmed by cell counts performed on formalin-fixed aliquots obtained from the 2 lowest dilutions of T. foetus (i.e., 10,000 and 1,000 organisms). DNA extractions were performed as directed by the manufacturer with the exception of kit B, where a previously optimized protocol 3 in which an extended incubation (56°C) of the extracted DNA with a higher concentration of proteinase K and an additional wash of the DNA was performed. For kit D, extraction of DNA from 100 mg of feces was also performed subsequent to consultation with the manufacturer. DNA was eluted in 300 μl (kit A), 200 μl (kit B), 50 μl (kit C), or 100 μl (kit D) of the respective elution buffer. The presence of endogenous PCR inhibitors in each DNA sample was assessed as previously described by PCR amplification of a 876 base-pair fragment of bacterial 16S ribosomal RNA (rRNA) gene. 4 Each sample was then tested by using a single-tube nested PCR for amplification of the partial ITS1, 5.8S, and ITS2 rRNA genes of T. foetus. 3 PCR amplification of bacterial 16S rRNA genes was observed for all fecal samples extracted by using kits C and D (41 each). Failure to amplify 16S rRNA genes was observed in 11 of 41 samples extracted with kit B (27%) and 1 of 41 samples extracted with kit A (2%). PCR performed on DNA extracted from feces (150 mg) by using kit D was able to detect as few as 10 T. foetus per fecal sample in 1 of 3 cats. Reproducibility (i.e., the ability to repeatedly detect a given number of T. foetus organisms, irrespective of the source of the feline feces) of kit D for detection of 10 T. foetus per fecal sample was improved to 3 of 3 cats by reducing the amount of feces (100 mg) used in the extraction (Table 1). For kit D, determination of analytical sensitivity of PCR for amplification of T. foetus was performed by using DNA extracted from 20- and 120-μl aliquots that contained 1, 10, 100, or 1,000 T. foetus in the presence (20 μl) or absence (120 μl) of 100 mg of feline feces, respectively. In the absence of feces, PCR detected ≥100 organisms per extraction in 100% of reactions (10/10 at each dilution), 10 organisms per extraction in 90% of reactions (9/10), and 1 organism per extraction in 40% of reactions (4/10). In the presence of feces, PCR detected 10 organisms per extraction in 100% of reactions (9/9) and 1 T. foetus organism per extraction in 0% of reactions (0/10). The similar analytical sensitivity of PCR by using DNA directly extracted from T. foetus dilutions compared with T. foetus-spiked feces (100 mg) suggests effective removal of PCR inhibitors when using kit D.
Number of cats from which fecal DNA tested positive for polymerase chain reactions (PCR) amplification of Tritrichomonas foetus partial ribosomal RNA gene unit.*
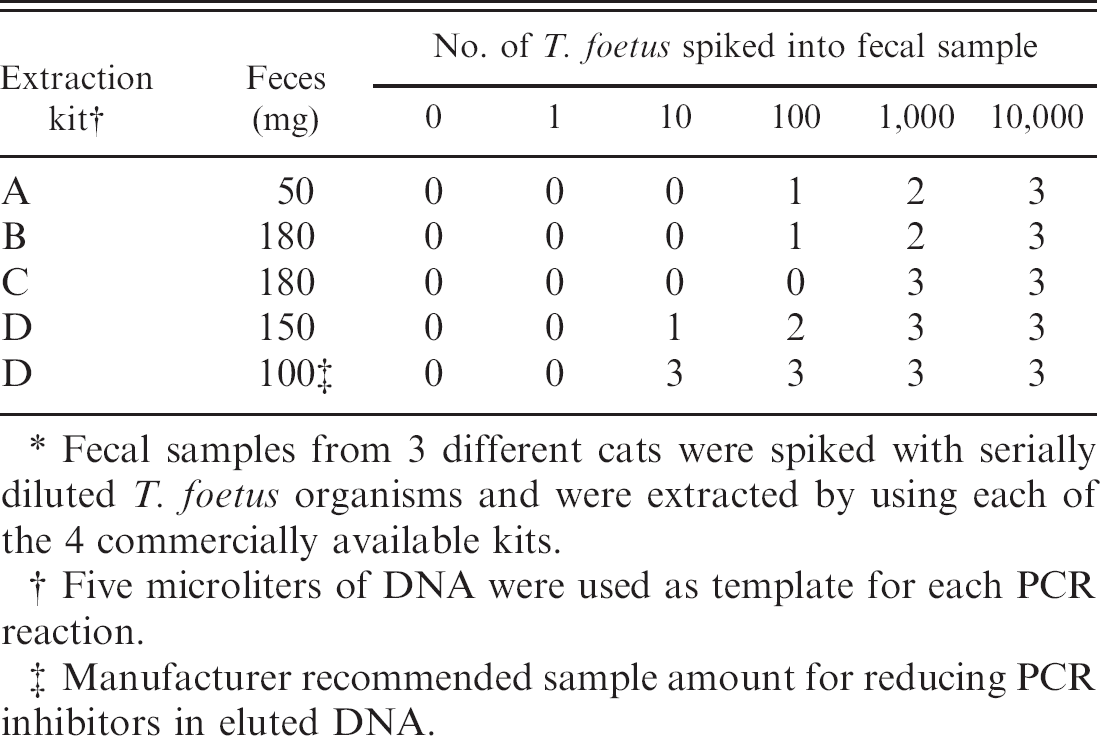
Fecal samples from 3 different cats were spiked with serially diluted T. foetus organisms and were extracted by using each of the 4 commercially available kits.
Five microliters of DNA were used as template for each PCR reaction.
Manufacturer recommended sample amount for reducing PCR inhibitors in eluted DNA.
Although the purpose of this study was to compare the performance of each kit as recommended by the manufacturer, each kit differed in the quantity of feces and elution buffer, which resulted in different concentrations of total DNA in each extraction. For each eluate (n = 90), 10 μl DNA was diluted in 40 μl of nuclease-free water or 40 μl Tris-chloride buffer (10 mmol, pH 8.5) for measurement of spectrophotometric f absorbance at wavelengths of 260 and 280 nm, respectively. Measurements were performed in triplicate, averaged, and used to calculate the DNA concentration (A260 in ng/μl) and purity (A260/280). The average concentration of DNA (ng/μl ± SD) obtained per fecal sample from each kit (n = 18 each) was as follows: kit A = 11.9 ± 5.2, kit B = 38.3 ± 7.6, kit C = 31.6 ± 9.0, kit D (150 mg) = 59.4 ± 6.2, and kit D (100 mg) = 50.3 ± 8.5. The average purity of DNA (A260/280 ± SD) obtained per fecal sample from each kit (n = 18 each) was as follows: kit B = 1.2 ± 0.3, kitC = 1.4 ± 0.5, kitD (150 mg) = 1.2 ± 0.1, and kit D (100 mg) = 1.0 ± 0.2. For kit A, absorbance at 280 nm was below the detection limit of the spectrophotometer. Spiking feces with T. foetus (1–10,000 organisms/sample) did not substantially alter the total DNA concentration or purity obtained by using any of the kits. Interestingly, the purity of the DNA as assessed by A260/280 did not translate into improved PCR sensitivity, perhaps because fecal proteins are not a major cause of PCR inhibition.
Number of positive polymerase chain reaction reactions versus total samples tested for ribosomal RNA genes by using DNA (250 ng) extracted from fecal samples of 3 cats spiked with 10 Tritrichomonas foetus by using kits.
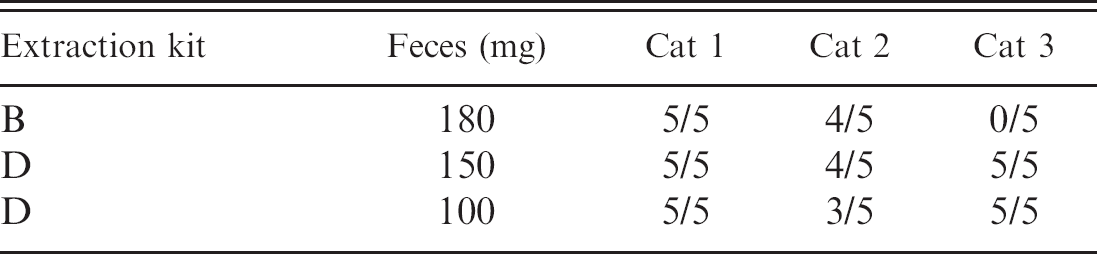
To determine if differences in the sensitivity and reproducibility of PCR among the kits simply reflected variation in the concentration of DNA obtained, we performed 5 replicate PCR reactions for each of 3 cats by using 250 ng of template DNA from T. foetus-spiked feces (10 organisms) extracted by using kit B and kit D (150 mg and 100 mg). Insufficient quantities of DNA were available for inclusion of kits A and C in these studies. By using equal quantities of DNA, T. foetus was detected by PCR in the feces of all 3 cats by using kit D (100 or 150 mg of feces) but only 2 of 3 cats by using kit B (Table 2). Thus, increasing the amount of DNA from kit B used in the PCR reaction improved the sensitivity but not the reproducibility of the assay. Poor reproducibility of kit B was also observed in this study as the failure to demonstrate the presence of 16S rRNA genes in 27% of DNA samples. After normalizing for DNA concentration, kit D was both sensitive and reproducible for demonstration of T. foetus rRNA genes by PCR, regardless of the quantity of feces used in the extraction (100 or 150 mg). This finding suggests that the greater concentration of DNA and/or PCR inhibitors extracted from 150-mg fecal samples by using kit D may result in poor reproducibility when ≥5 μl of eluate is used directly in PCR.
To assess the performance on clinical samples from naturally infected cats, DNA was extracted in accordance with manufacturer instructions from diarrheic feces of 20 cats from which samples were submitted by referring veterinarians for PCR testing for T. foetus. Ten positive samples and 10 negative samples were selected based on PCR results obtained by using DNA extracted from 100 mg of feces by using kit D. For the remainder of each fecal sample, 180 mg (kits A and B) and 50 mg (kit C), aliquots were extracted by using the respective kits. An equal volume of extracted DNA from each kit (5 μl) was added to each PCR reaction. There was 100% concordance in PCR results obtained (i.e., 10 positive and 10 negative samples) by using DNA extracted by kits A, B, and D. For kit C, only 9 of 10 positive samples were correctly identified. Notably, DNA extracted from kits A and B failed to yield amplicons from a simultaneously run positive control extraction that contained 100 mg of feces spiked with 1,000 T. foetus. In contrast, DNA extracted from the positive extraction control by using kit D resulted in a robust PCR product, whereas only a faint PCR product was amplified from the positive control extraction by using kit C. These results suggest that the clinical samples from cats with symptomatic T. foetus infection (i.e., diarrhea) contained sufficient numbers of T. foetus (likely ≥10,000 organisms) to be detected in DNA extracted when using any one of the kits. However, differences in ability to detect T. foetus (1,000 organisms) in the positive control extraction agreed closely with the predicted sensitivity data for each kit as shown in Table 1. Accordingly, kit D would be predicted to enable the detection of greater numbers of T. foetus infected fecal samples compared with other kits if a larger number of clinical samples (that contain a broader range in numbers of T. foetus) were tested. Future studies should include the screening of clinical samples from cats without diarrhea that may be carriers of the infection with lower numbers of parasites.
Comparison of cost per sample, average length of procedure, and technical aspects of performance for 4 commercially available kits for extraction of DNA from fecal specimens.

Investigator average length of procedure based on the simultaneous extraction of 13 fecal samples.
Does not include optimized conditions (i.e., 1-hr incubation in proteinase K and extra DNA wash).
In addition to assessing sensitivity and reproducibility, cost per sample, average length of procedure, and technical aspects of performance (subjectively assessed as good or fair) for each kit were compared (Table 3). Although kit D extracted PCR-quality DNA in the shortest period of time, the authors did not consider the kit among the most user-friendly. This was primarily because of the use of screw-top rather than snap-top caps.
The sensitive detection of T. foetus DNA by PCR in fecal samples extracted with kit B with minor modifications was previously described. 3 However, a decrease in sensitivity that appeared to correspond to a change in the components of the kit was detected in the authors' laboratory. This prompted investigations to determine the optimal DNA extraction kit for detection of pathogen DNA in feline feces. Indeed, DNA extracted from feces by using kit B, and used directly in PCR, was considerably less sensitive and reproducible for detection of T. foetus than previously reported. Although the sensitivity of kit B was improved by adding more DNA to the PCR reaction, failure to reproducibly demonstrate bacterial 16S rRNA and T. foetus rRNA genes remained a problem. In comparison, kit D provided reproducibly sensitive detection of T. foetus rRNA and bacterial 16S rRNA genes in every sample.
Footnotes
a.
ExtractMaster Fecal DNA Extraction kit, Epicentre Biotechnologies, Madison, WI.
b.
QIAamp DNA Stool Mini kit, Qiagen, Valencia, CA.
c.
UltraClean Fecal DNA kit, MoBio, Carlsbad, CA.
d.
ZR Fecal DNA kit, Zymo Research, Orange, CA.
e.
Hill's Science Diet Maintenance, St. Louis, MO.
f.
Eppendorf Biophotometer, Hamburg, Germany.