
Abstract
This document is the consensus of the American Association of Veterinary Laboratory Diagnosticians (AAVLD) Subcommittee on Standardization of Immunohistochemistry on a set of guidelines for immunohistochemistry (IHC) testing in veterinary laboratories. Immunohistochemistry is a powerful ancillary methodology frequently used in many veterinary laboratories for both diagnostic and research purposes. However, neither standardization nor validation of IHC tests has been completely achieved in veterinary medicine. This document addresses both issues. Topics covered include antibody selection, fixation, antigen retrieval, antibody incubation, antibody dilutions, tissue and reagent controls, buffers, and detection systems. The validation of an IHC test is addressed for both infectious diseases and neoplastic processes. In addition, storage and handling of IHC reagents, interpretation, quality control and assurance, and troubleshooting are also discussed. Proper standardization and validation of IHC will improve the quality of diagnostics in veterinary laboratories.
Introduction
Morphological diagnosis in veterinary medicine has classically relied mostly on routine stains such as hematoxylin and eosin and, less commonly, on other histochemical stains. However, the level of specialization in veterinary practice demands more accurate diagnosis, particularly for tumors. Immunohistochemistry (IHC) has been proven to be one of the most important ancillary techniques in the characterization of neoplastic diseases in humans and has become equally important in veterinary medicine, as oncologists demand more specific diagnoses. The number of immunohistochemical tests offered by veterinary diagnostic laboratories for the diagnosis of infectious and neoplastic diseases in frozen or formalin-fixed, paraffin-embedded (FFPE) tissues has increased exponentially in the last decade. Immunohistochemistry has been proven as a highly specific and sensitive diagnostic method that is especially advantageous as a diagnostic tool for infectious diseases. In some cases, IHC is considered the gold standard technique to which others are compared (e.g., prion diseases). In comparison with other diagnostic tests, IHC allows colocalization of an antigen with a lesion, thereby dramatically increasing diagnostic accuracy and understanding of pathogenesis. Numerous infectious agents and cell types can be identified with IHC in a wide variety of animal species, especially since the advent of antigen retrieval methods using heat. 62 However, there are no guidelines for standardization and no general agreement on the use of controls or the validation of a test in veterinary diagnostic IHC. The purpose of the present article is to provide guidelines for IHC in the veterinary diagnostic laboratory, and is not intended as mandatory requirements for laboratory accreditation. The article briefly reviews the technical aspects of IHC and provides guidance for standardization, test validation, controls, storage and handling, interpretation and quality control and assurance, as well as general troubleshooting protocols.
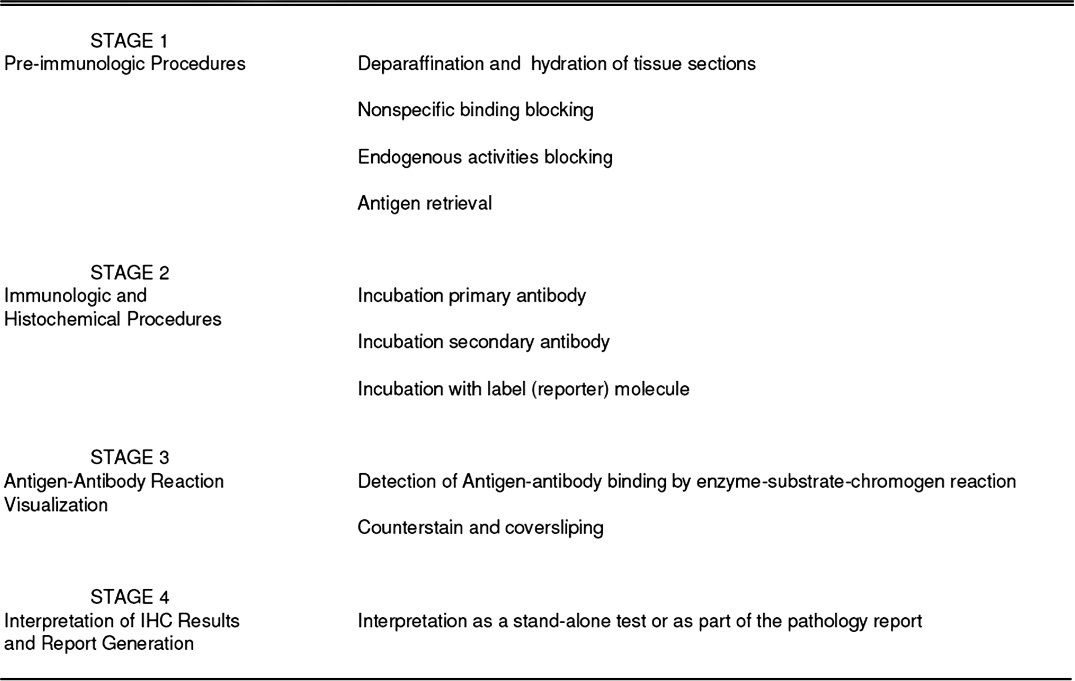
Overview of immunohistochemical test. The immunohistochemistry technique can be divided into 4 main sections: pretreatments (stage 1), immunologic and histochemical reactions (stage 2), visualization of the immunologic reaction (stage 3), and interpretation and report generation (stage 4).
Overview of the immunohistochemical test
The IHC technique is a combination of immunologic and chemical reactions visualized with a photonic microscope. The IHC technique can be divided into 4 main steps (Fig. 1). Step 1 includes all preimmunologic procedures done before incubation with the primary antibody. These procedures include deparaffinization and hydration of tissue sections, blocking nonspecific bindings, blocking endogenous activities (e.g., peroxidase, phosphatase, biotin), and antigen retrieval. Step 2 includes all the immunologic reactions between the primary antibody and tissue antigens, the primary antibody and secondary antibody and some additional chemical reactions necessary to bind the reporter molecule (label) such as peroxidase or alkaline phosphatase to the preformed immune complex. Step 3 includes all the procedures necessary to visualize the antigen-antibody binding. In IHC, this is achieved with a chemical reaction in which the label reacts with its substrate and a chromogen to produce a colored reaction product. Then, immunolabeled tissue sections are counterstained and coverslipped. Step 4 includes interpretation and reporting of the IHC results.
Standardization of a new test
Standardization determines optimal conditions (e.g., incubation time, incubation temperature, dilutions, controls, buffers, detection system) to ensure that the selected antibody reacts with the expected antigen (Fig. 2). There is a lack of standardization among different veterinary laboratories, despite the widespread use of IHC. 47 Contrary to the situation in human IHC, a major problem in veterinary IHC is the reduced availability of high-quality antibodies that are specific for infectious agents of veterinary importance, and cell markers specific for small and large animal species. Especially in tumor diagnostics, most laboratories use batteries of antibodies developed for human tissues that are often insufficiently validated for animal species. With the exception of the surveillance program for prion diseases (e.g., scrapie, chronic wasting disease), neither the protocols nor the primary antibodies are uniform among diagnostic laboratories.
There is also little guidance or regulation for the actual standardization of specific IHC tests in human medicine although a consensus on guidelines for quality assurance for IHC has been published by the Clinical and Laboratory Standards Institute (formerly National Committee on Clinical Laboratory Standards, 1999). 1 The Food and Drug Administration (FDA) has classified most IHC reagents and kits as “Analyte Specific Reagents,” thereby exempting them from premarket notification (FDA regulations classify devices depending upon the degree of regulation necessary to provide reasonable assurance of their safety and effectiveness). This ruling was based on the assumption that most IHC tests are part of the pathologist's report and, as such, will not be reported independently. For antibodies used as stand-alone tests in human pathology, premarket notification and specific FDA approval are required. For stand-alone tests in veterinary IHC, premarket notification by regulatory agencies (U.S. Department of Agriculture [USDA]) is not required. However, for certain animal diseases (e.g., scrapie, chronic wasting disease, Bovine viral diarrhea virus), IHC results are commonly reported as stand-alone tests, so standardization of these tests among various laboratories is urgently needed. The main goal of standardization in IHC is to obtain reproducible and consistent results within each laboratory and comparable results among different laboratories even when using different procedures. In this review, each of the main components of an IHC test, including primary antibody, fixation, antigen retrieval, pretreatments, and detection systems, are addressed.
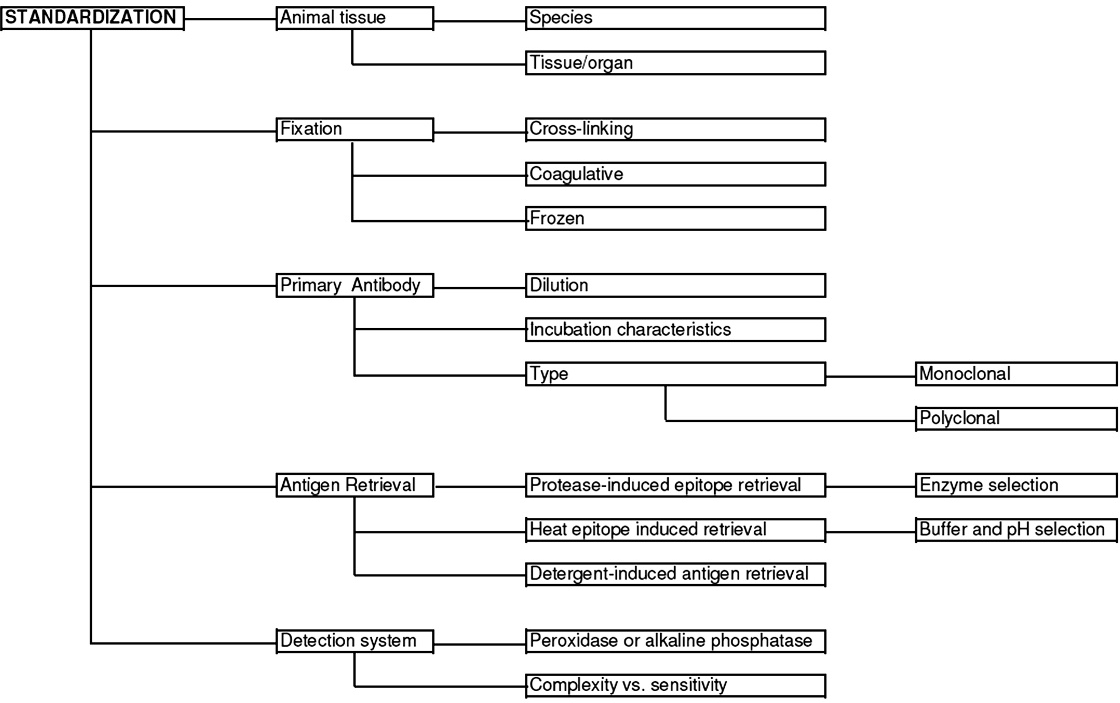
Steps to consider during the standardization of a new immunohistochemical test. Five main factors are involved in immunohistochemistry standardization: animal tissue, fixation, primary antibody, antigen retrieval, and detection systems.
Primary antibody characterization
Monoclonal and polyclonal antibodies: sensitivity and specificity. Monoclonal antibodies (mAb) derived from a single B-cell clone and produced by hybridoma technique provide excellent specificity because the antibody binds to a single epitope on 1 antigen. 43 most mAb are of mouse origin, although rabbit mAb are also commercially available. 56,69 Polyclonal antibodies (pAb) contain antibodies to a range of antigens and thus may cause greater nonspecific background staining and be less specific than mAb. Subjecting pAb to multiple adsorption protocols for a variety of antigens will increase their specificity. For both mAb and pAb, the diagnostic specificity of an antibody detecting infectious agents is the positive immunoreactivity against the targeted agent only and lack of immunostaining in tissues infected with other agents. The diagnostic specificity of an antibody detecting cellular/tumor markers is the expected presence or absence of immunostaining in certain cell types, tissues, or tumors in multitissue control blocks (see Controls section). 64 Analytical sensitivity is defined as weak (positive) staining in the presence of the least amount of antigen. Polyclonal antibodies may be more sensitive than mAb because they can bind several different epitopes on a single antigen. 64 Diagnostic sensitivity, defined as the proportion of known positive samples that test positive, 65 is established by comparing the test results on FFPE tissue with results using another antibody that has been validated for the same analyte or using a non-IHC method such as culture or polymerase chain reaction (PCR).
Cellular markers.—With the exception of CD (cluster of differentiation) markers, there are few cellular antigens in animals for which species-specific antibodies have been developed, and even fewer are detectable in FFPE tissues. Most antibodies to cellular antigens used in veterinary IHC laboratories have been developed against human or rodent antigens, and characterization data for their specified use should be available. Cellular markers should demonstrate reactivity with the appropriate molecular weight antigen in Western blots (WB); however, WB immunoreactivity does not necessarily imply or predict immunoreactivity in FFPE tissues.
Interpretation of IHC results requires familiarity with the expected pattern of immunoreactivity based on location of the antigen (e.g., nuclear, membrane, or cytoplasmic staining; Fig. 3). 76 For example, staining for cytokeratins and vimentin should be cytoplasmic, not nuclear; staining for thyroid transcription factor 1 (TTF 1) in lung or thyroid tumors is nuclear, not cytoplasmic. 48,51 In other words, the presence of staining does not always indicate a positive reaction. In most cases, the degree of specific staining should vary between cells and in some cases within different cell compartments; such cellular distribution may have prognostic significance in certain tumors. 33
Infectious agents.—For viral agents, positive staining should be detected in the appropriate tissues, cell types, and cellular location (if known), in association with typical lesions. For protozoal and bacterial pathogens, the morphology and location of the stained organisms can provide some additional information.
Fixation
The ultimate goal of fixation is to preserve cells and tissues, to prevent autolysis, and to preserve antigenicity. Unfortunately, no fixative can optimally fulfill all these requirements. 43 The most common type of fixation in diagnostic IHC is chemical, specifically with formaldehyde. The time lapse between the death of the animal and the collection of tissues for IHC is usually critical, and prompt transfer into fixative is desired. Enzyme-rich tissues, such as intestine or pancreas, autolyze rapidly. Protozoa and fungi may be more resistant to autolysis than viruses. In general, samples to be fixed should not be thicker than 0.5 cm, and the ratio of fixative to sample should be at least 10 to 1. Factors influencing the quality of fixation include type of fixative, fixative pH and buffers, fixative concentration, fixative osmolality, fixation time, fixation temperature, fixative additives, and the use of additional post fixation procedures (e.g., decalcification).
Coagulative fixation. Coagulative fixatives are organic and nonorganic solutions that coagulate proteins and render them insoluble. The fixation process removes and replaces free water, thereby destabilizing hydrophobic and hydrophilic bonding (hydrophobic areas are released from the repulsion of water, and occupy a greater area). This causes a disruption of tertiary protein structure and results in a partially reversed (“less folded”) protein structure. 43 The disruption of tertiary structures also changes physical properties, and proteins that are normally soluble in aqueous solution become insoluble.
Coagulative fixation maintains tissue structure at the light microscopic level fairly well, but results in cytoplasmic flocculation as well as poor preservation of mitochondria and secretory granules. 43 The most common types of coagulative fixatives are dehydrants (alcohols and acetone) and strong acids (picric acid, trichloroacetic acid).
Cross-linking fixation. Cross-linking fixatives form cross-links within and between proteins, within and between nucleic acids, and between nucleic acids and proteins. The most common cross-linking fixatives are aldehydes (formaldehyde, glutaraldehyde, chloral hydrate, glyoxal), with neutral buffered formaldehyde most frequently used in routine histopathology. Formalin fixation produces methylene bridges between free amino groups and other functional groups of proteins, resulting in conformational changes in the tertiary and quaternary structures within proteins and cross-linking between tissue proteins, thus modifying the epitopes recognized by antibodies. 3,15,21,22,43,71 However, it needs to be emphasized that adequate fixation is essential in IHC. Underfixation of tissues for IHC testing is one of the major problems in human pathology. 15 Tissues that are poorly fixed in formalin will be subjected to coagulative fixation (alcohol) during tissue processing, resulting in inadequate antigen retrieval or unexpected antigen detection. Underfixed tissues cannot withstand harsh retrieval methods and are easily damaged, with subsequent loss of antigenicity. 54
Antigen retrieval
Antigen retrieval (AR) is intended to reverse the detrimental effects of fixation. As mentioned, one of the main effects of formalin fixation is conformational changes in epitopes, but loss of electrostatic charges may occur during fixation and have an effect on antigen detection. 16 The exact mechanism by which AR works on formalin-fixed tissues is not clear. A variety of pathways may contribute to its success, including the breaking of cross-linkages, the extraction of diffusible blocking proteins, the precipitation of proteins, the hydrolysis of Schiff's bases, calcium chelation, paraffin removal, and the rehydration of tissue, resulting in better penetration of antibody and increased accessibility to antigen. 37,62 Longer fixation times are thought to decrease immunoreactivity and might require longer, harsher AR methods, increased antibody concentrations or longer primary antibody incubation times to achieve a similar degree of immunoreactivity. However, with the advent of heat-induced AR, retrieval of antigens in overfixed tissues can still be achieved in many instances. 45,48,51 In a recent study using 30 antibodies targeting cellular antigens in different cell compartments and infectious agents, only those to Canine parvovirus, Felid herpesvirus, and cytokeratin 7 had a significant reduction in the signal after several weeks of fixation (Webster J, Miller M, Ramos-Vara J: 2007, Proc AAVLD, p. 86). Background staining caused by harsh AR methods is not uncommon, and sometimes precludes diagnostic evaluation of the staining. Three main AR methods are currently used in FFPE tissues: heat-induced AR, enzymatic digestion, and detergent-induced AR (Fig. 4). For a practical approach to antigen retrieval, see the next section (“Practical approach to standardization of a new antibody”).
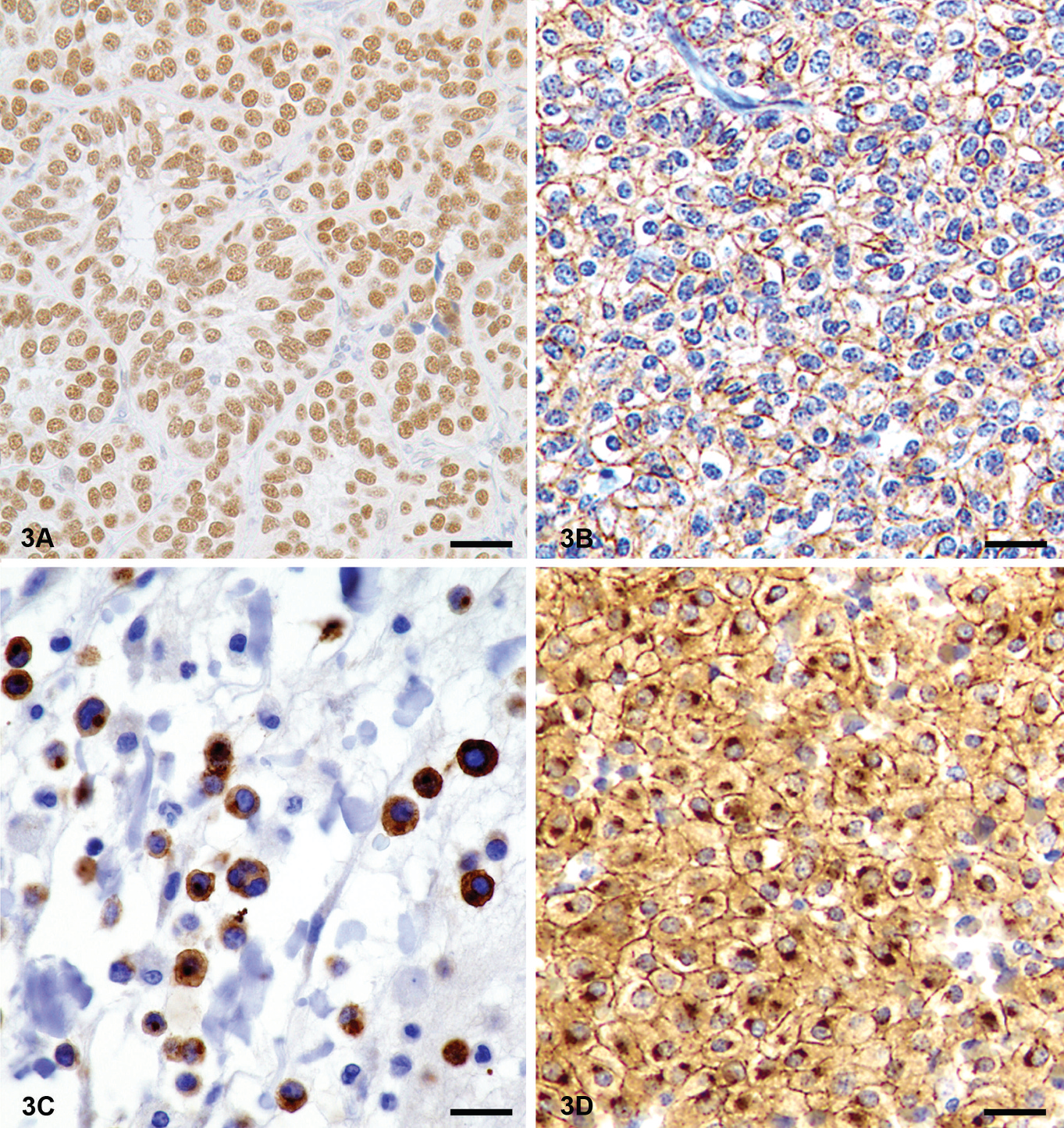
Cellular location of antigens. Antigens can be located in one or multiple cell compartments. Knowledge of the expected location of a particular antigen is essential to an adequate interpretation of the immunohistochemistry results.
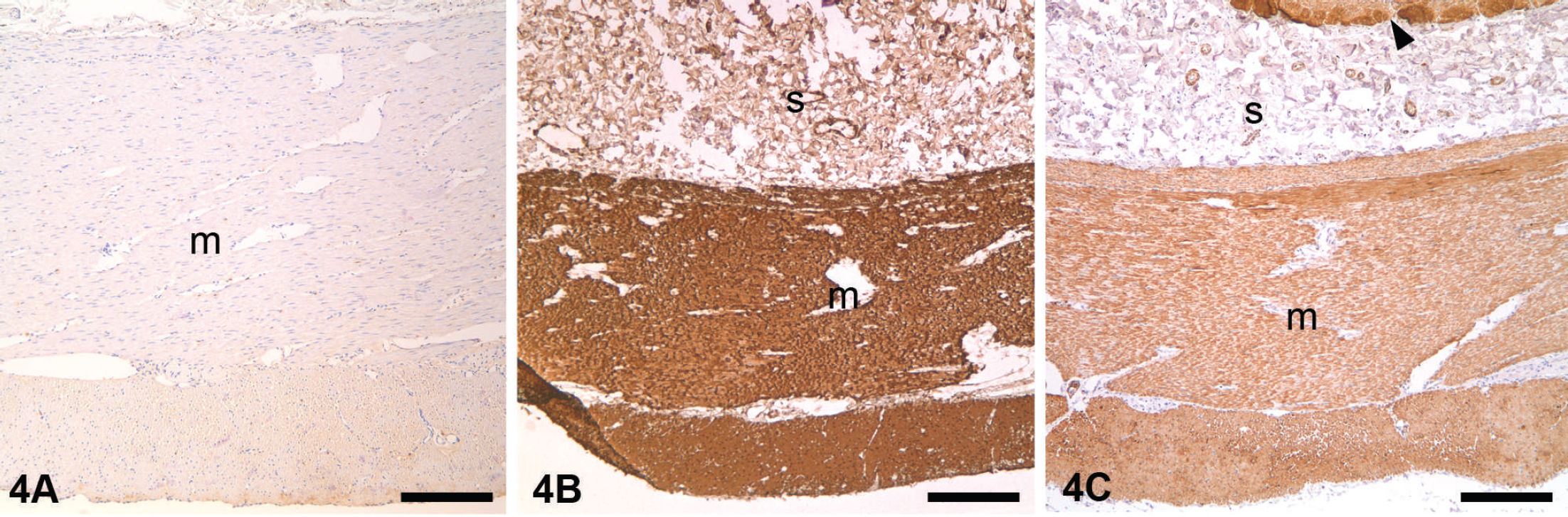
Effects of different antigen retrieval (AR) in the immunohistochemical reaction. Small intestine, dog.
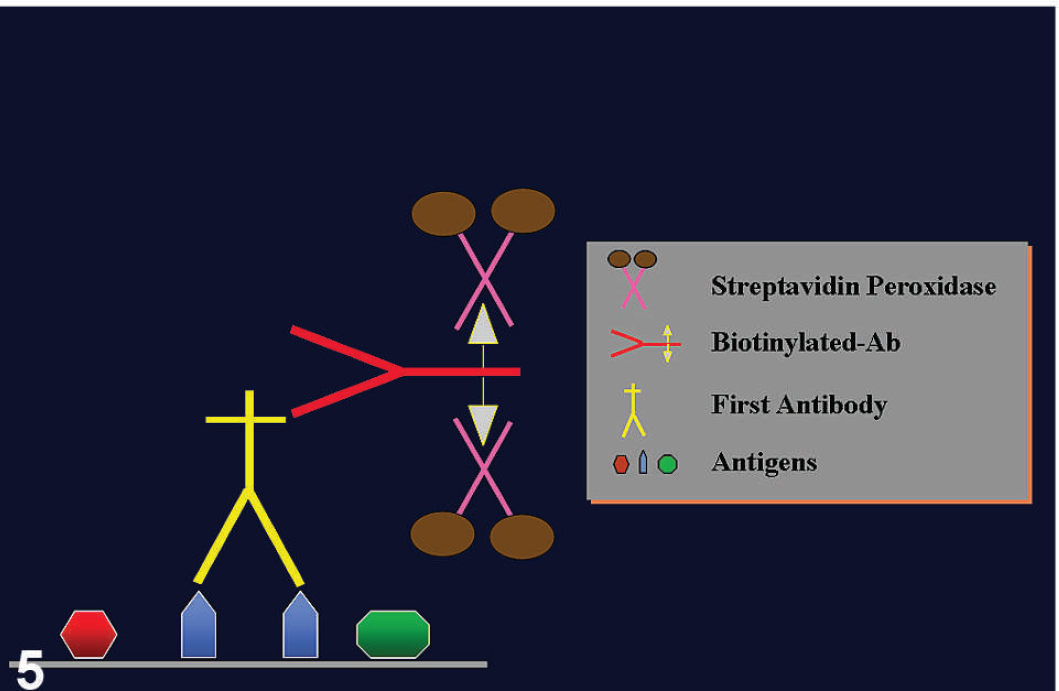
Labeled streptavidin–biotin peroxidase method. This 3-step method consists of primary antibody-binding tissue antigens, a secondary biotinylated antibody recognizing the primary antibody, and avidin–peroxidase complexes that will bind the secondary antibody via avidin–biotin bonds (reprinted from Ramos-Vara 43 with permission from Allen Press).
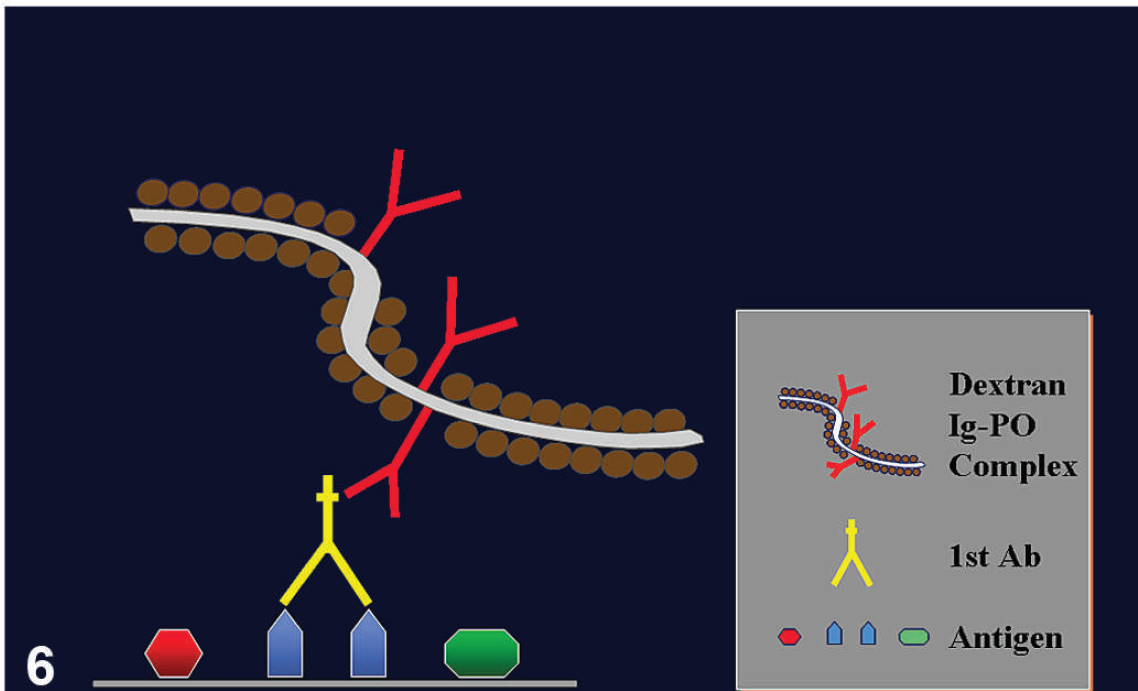
Polymer-based technology. This is a 2-step method. The primary antibody will bind tissue antigens. The immunoglobulin–peroxidase–polymer complex will bind via immunoglobulins to the primary antibody. This method does not involve avidin and biotin molecules (reprinted from Ramos-Vara 43 with permission from Allen Press).
Heat-induced antigen retrieval
The success of heat-induced AR is probably the result of reversal of formalin-induced chemical modifications in protein structure. The source of heat is not critical to the outcome, but a matter of convenience. Various types of equipment may be used including a de-cloaker (commercial pressure cooker with electronic controls for temperature and time), vegetable steamer, microwave oven, or pressure cooker. 7,20,32,41,63 Many veterinary diagnostic laboratories use a steamer and/or de-cloaker. The advantage of a de-cloaker over other heating devices is that the boiling temperature is not affected by the atmospheric pressure, which varies depending on the altitude.
The relationship between temperature and exposure time is inverse: the higher the temperature, the shorter the time needed to achieve beneficial results. The pH of the retrieval solution is important. 62 Some antibodies bind well regardless of retrieval solution pH, whereas others bind weakly at neutral pH, but strongly at very low or high pH. Common buffers used in heat-induced AR are citrate, Tris-HCl, and EDTA (ethylenediamine tetra-acetic acid). A low pH buffer (acetate, pH 1.0–2.0) appears especially useful for nuclear antigens. The significance of the chemical characteristics of the retrieval solution, particularly the buffer, is unclear.
Enzymatic digestion. Enzymes are thought to digest the tissue to some degree, allowing antibodies to recognize antigenic sites. This digestion is nonspecific and some antigens might be negatively affected by this treatment. 68 Commonly used enzymes include trypsin, proteinase K, pepsin, pronase E, and ficin. A combination of heat and protein digestion may be necessary to demonstrate some antigens.
Detergent-induced antigen retrieval. Detergents solubilize membrane proteins by mimicking the lipid-bilayer environment. 54 They form mixed micelles that consist of lipids and detergents as well as detergent micelles that contain proteins (also known as surfactants), thereby decreasing the surface tension of water. They are usually incorporated into the dilution/rinse buffers. Examples of detergents include ionic detergents, bile acid salts, and nonionic and zwitterionic types (e.g., Triton R-X 100, BRIJR, Tween 20, saponin).
Practical approach to standardization of a new antibody
Dilutions of primary antibody. Antibody dilutions for one type of tissue may not work with different tissues for the same antigen (e.g., brain vs. lymphoid tissue). Differences in processing techniques and other variables may require antibody titration for each tissue type. Often the manufacturing company has data on antibody performance in FFPE tissue that can be used as a guideline for the starting dilution. Typically 2- to 10-fold dilutions above and below the manufacturer's recommended dilution provide a good starting point. Depending on the antibody type (monoclonal or polyclonal) and concentration, a range of 1–5 μg/ml should be used for an initial titration. The optimal working concentration for most antibodies is generally from below 1 μg/ml to 3 μg/ml, depending on the detection system utilized.
If an antibody has not yet been tested, a systematic approach using a wide range of dilutions might be needed, as well as prolonged (e.g., overnight) incubations with the primary antibody. Secondary antibodies (linker reagents) and detection complexes in commercially available detection kits do not need to be titrated. For practical purposes, when testing a new antibody, it is recommended to use 3 sets of 5 slides (15 slides total): one set without AR, one set with enzymatic digestion, and one set with heat-induced AR. The first 4 slides in each set will have 2-fold dilutions of the primary antibody and the fifth slide should be incubated with the negative reagent control. 44,54 A similar approach has been suggested by the College of American Pathologists. 28
Incubation conditions. The conditions for incubation of the primary antibody depend on the antibody characteristics (e.g., affinity), environmental factors (e.g., temperature), section characteristics, and procedure. Incubation for most routine IHC protocols is 30–90 min at room temperature. During incubation, the sections must be completely covered by an adequate volume of the antibody solution to prevent section drying and assure even exposure of the tissue to the antibody solution. Shortened incubation times at 37°C, longer incubation times at room temperature, or extended overnight incubation in a humidity chamber at 4°C is occasionally utilized to achieve adequate immunostaining. 17 Additional tissue treatments (e.g., decalcification), length of fixation, and AR technique can affect incubation of the primary antibody. Decalcification with weak acids does not seem to interfere significantly with immunostaining of most antigens, provided the tissues were previously well fixed in formaldehyde. Strong acids may affect the detection of some antigens at various degrees. 2,4,35 It is recommended to use weak acidic decalcifying solutions (e.g., formic acid) diluted in formalin.
Primary antibody characteristics.—There are many characteristics of the antibody that affect incubation time. The specificity of the antibody is important in determining the length of incubation time to get sufficient binding. Lower-affinity antibodies require longer incubation times (and/or higher concentrations). Affinity rarely has to do with whether the antibody is monoclonal or polyclonal. Other antibody-dependent factors include immunoglobulin isotype, manufacturer, clone, and lot differences. Different lots of the same antibody may vary in concentration or other solution characteristics and thus require re-evaluation.
Environmental factors.—Incubation temperature is a major determinant of incubation time. In general, as temperature increases, incubation time decreases. Some antibodies might be temperature-sensitive with decreased immunoreactivity at higher temperatures. Sections with longer incubation times may dry out if there is inadequate humidity in the incubation chamber. Performing the incubation in humidity chambers helps to alleviate this complication.
Tissues from different animal species.—Species of origin of the tissue can dramatically affect reactivity. Interspecies variations in antibody reactions results from subtle changes in the amino-acid sequence of a given antigen in a particular species. Even in the event of interspecies cross-reactivity, the antibody affinity may be decreased. Prolonged incubation times, varied AR methods, and/or increased antibody concentration may be needed to obtain optimal staining. Specificity of the antibody must be verified for every species tested.
Preservation procedure. The type of tissue preservation will likely affect the performance of the primary antibody. Frozen sections typically require less incubation time than do FFPE tissue sections. Alcohol or acetone fixation has variable effects on the incubation characteristics of the primary antibody. There are commercially available fixatives that reportedly preserve epitopes better for IHC techniques than the standard formaldehyde, 43 but these fixatives can be more expensive and less suitable for routine microscopy.
Choice of detection system. The sensitivity of IHC tests depends mainly on the detection system used. As a general rule, the more complex an IHC method, the more sensitive it is. One step or 2-step IHC procedures are usually less sensitive than more complex, multistep procedures. Most detection systems currently used in diagnostic laboratories are based on a color change induced by an enzyme attached to the immuno-complexes bound to a tissue section, after reacting with its substrate and chromogen. The 2 most common detection systems are avidin–biotin and polymer-based nonavidin–biotin systems.
Avidin–biotin systems. These systems are based on the strong affinity of avidin for biotin. Avidin–biotin systems are currently the most commonly used detection systems. Biotin is usually attached to a secondary antibody that binds a complex of avidin–enzyme to produce a colored reaction. These methods are very sensitive (Fig. 5). Problems with endogenous biotin background, particularly when using harsh (heat) AR methods or staining tissues especially rich in biotin (e.g., liver), may occur; blocking is usually necessary but is also expensive.
Polymer-based nonavidin–biotin systems. These systems are usually 2-step procedures (can be increased to a 3-step procedure if more sensitivity is necessary; Fig. 6). The first step is the unlabeled primary antibody; the second step consists of a polymer containing numerous secondary antibodies as well as numerous molecules of enzyme. 59,75 These methods are faster than most current avidin–biotin–based methods, do not produce the background generated by endogenous biotin, and have comparable or sometimes superior sensitivity to avidin–biotin methods. They are also more expensive.
Additional detection systems. There are multiple variations to the above-mentioned systems aimed at increasing the sensitivity of the reaction. Examples of highly sensitive methods are the tyramine–biotin method (very sensitive but can create high background sometimes) and immunorolling circle amplification, which is a nonavidin–biotin method. 43,60 The most commonly used enzymes are peroxidase and alkaline phosphatase. 43,54,64
There are no rules of thumb in selecting a detection system. The choice of system will depend on several factors: 1) degree of expertise/experience of the technician; 2) type of antigens to be detected (some do not need very sensitive methods because they are abundant); 3) number of tests (antibodies) available (different antibodies may require different detection systems); 4) species idiosyncrasies (e.g., amount of endogenous biotin in tissues); 5) budget; and 6) best signal-to-noise ratio when combined with the AR method used.
Test validation
Validation of a diagnostic test is, in general, the process of optimizing the test method (reagents and protocols) and determining the performance characteristics of the test (Fig. 7). Requirements for test validation are published for AAVLD accreditation and provide useful general scientific definitions and practical steps for classifying laboratory tests as “validated for use,” but are not specific for IHC assays (Essential Requirements for an Accredited Veterinary Medical Laboratory Version 4.1, Section 5.4.3, 2006. Published by AAVLD). For most IHC assays, this involves detecting any cross-reactivity of the selected antibody with unrelated antigens, and cross-reactivity among different tissues and among different species. Moreover, validation examines the variables that affect the IHC reaction, such as fixation time and storage of paraffin blocks or storage of unstained tissue sections. Last, but not least, validation of an IHC test may include comparison of results among different laboratories using similar techniques. 9 When possible, validation compares the sensitivity of IHC detection to the gold standard method of detection for the antigen in question. It may also compare staining patterns and sensitivity with other antibodies targeting the same antigen. Validation is usually an ongoing process and generally requires extensive testing. Although the AAVLD IHC subcommittee believes that minimum standards for validation are needed, the exact validation procedure will depend on the selected antibody and the laboratory (in-house validation). As with standardization, one goal of validation is to produce the same result regardless of the IHC method used or the laboratory where the test is performed. The primary aim among laboratories should be documentation of their validation procedure(s) as it applies to their methods for the IHC detection of an antigen.
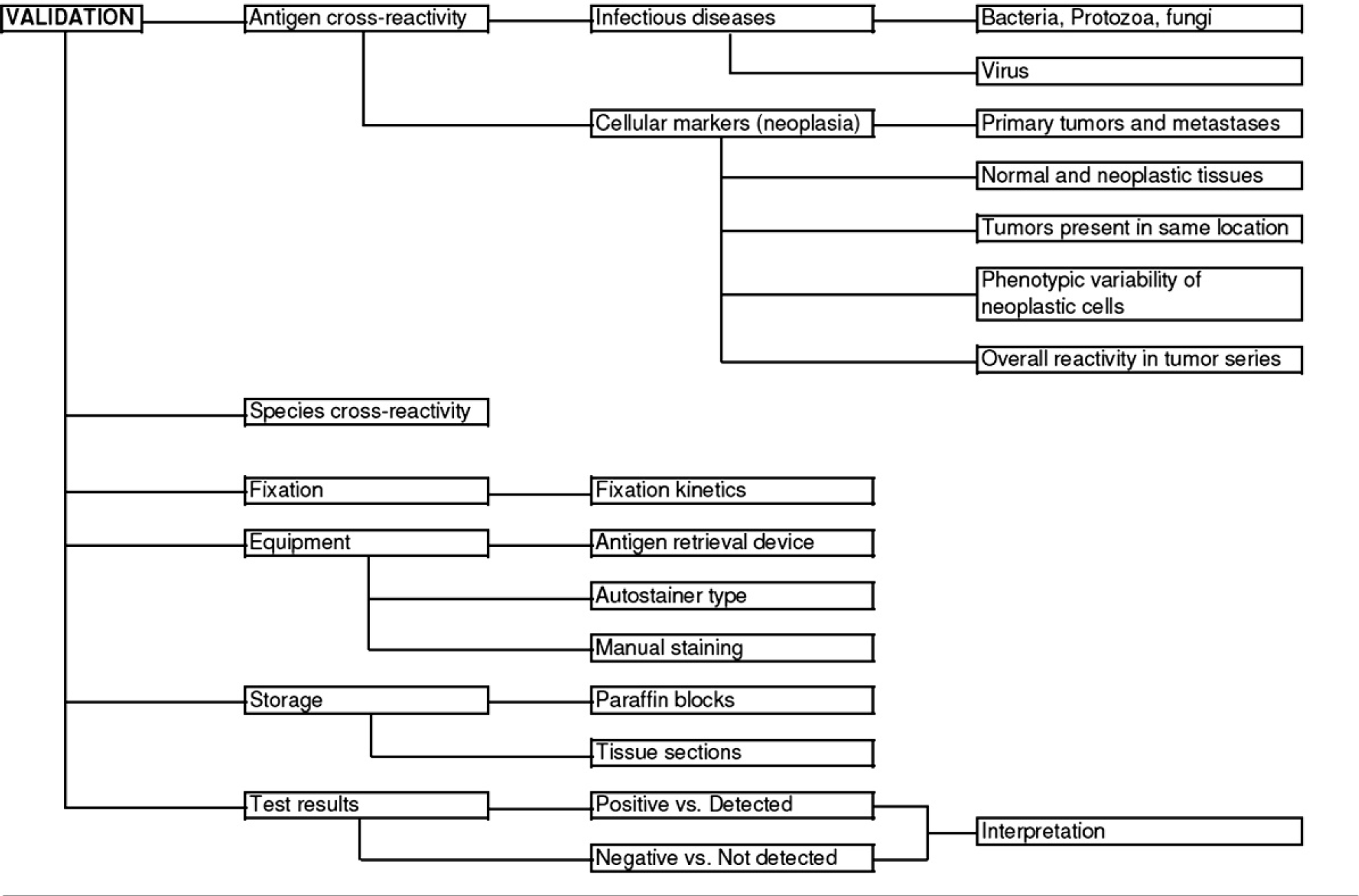
Validation of an immunohistochemical test. Factors to be included in the immunohistochemistry validation are antigen cross-reactivity, species cross-reactivity, fixation, equipment used, storage of reagents, and test results.
Cross-reactivity
Two main types of cross-reaction are considered: antigen cross-reactivity and species cross-reactivity. It is assumed that testing conditions (e.g., fixation, tissue processing, antigen retrieval, incubation times, detection kits) are the same as those used during standardization of the IHC test.
Antigen cross-reactivity. The type of antigen (i.e., structure, amino-acid sequence) affects cross-reactivity. A literature search may reveal reports of antigen cross-reactivity. Lack of cross-reactivity in 1 species does not preclude this potential problem in other species.
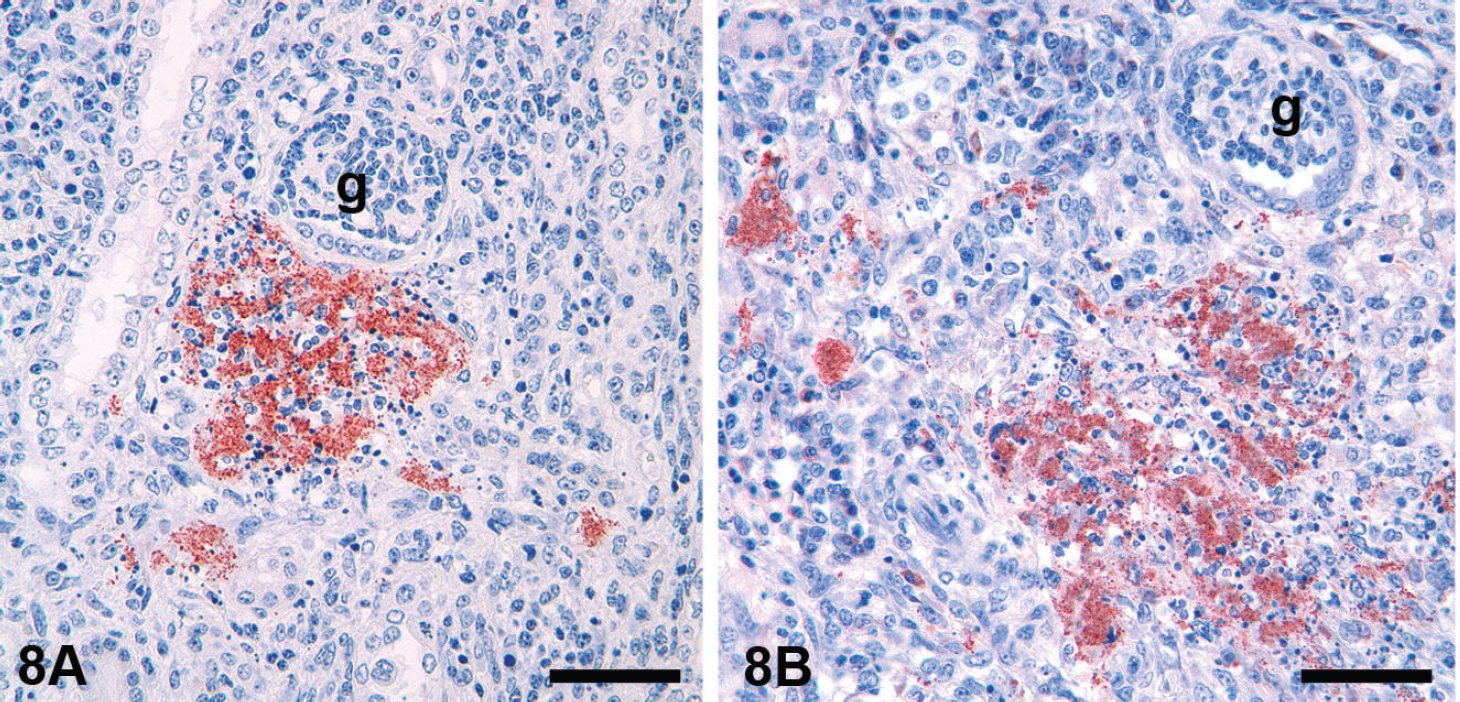
a) Infectious diseases.—For antibody-targeting bacteria, protozoa, and fungi, it is suggested that the cross-reactivity of an antibody to other bacteria/protozoa/fungi that are morphologically similar, belong to the same group, or produce similar lesions in the organ of interest, be determined. For example, an antibody to Leishmania sp. should be tested against other protozoa (e.g., Trypanosoma, Eimeria, Isospora, Cryptosporidium, Giardia, Neospora, Toxoplasma, Sarcocystis; Fig. 8) and morphologically similar fungi (e.g., Pneumocystis, Histoplasma, Sporothrix). For antibody-targeting viruses, it is necessary to test against other viruses within the same group that affect the same tissue or produce similar lesions. For example, an antibody to Canine parvovirus should be tested in tissues infected with Canine coronavirus, Canine distemper virus, and Canid herpesvirus.
Some lack of specificity does not necessarily preclude the use of an antibody provided it is documented and can be interpreted. An example of this would be the use of an antiserum raised against Measles virus nucleoprotein, which reacts with most known morbilliviruses, for the detection of Canine distemper virus in dogs. Some mAb against infectious agents will cross-react with cell organelles (Fig. 9).
b) Neoplastic diseases.—Developing IHC protocols for antibodies to specific cell types or cell components requires one or more of the following comparisons:
Comparison of immunoreactivity of primary tumors and their metastases. 45,48
Antibody cross-reactivity.
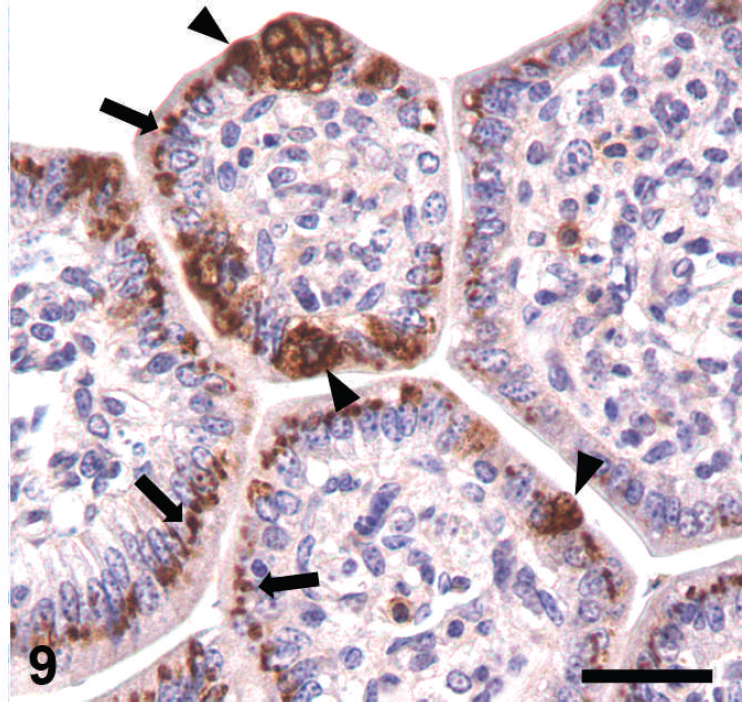
Antibody cross-reactivity. In this case, an antibody recognizing rotavirus (arrowheads) in a porcine intestine also cross-reacts with a supranuclear structure (interpreted as the Golgi apparatus) in most enterocytes (arrows). The authors have observed similar nonspecific reaction with other monoclonal antibodies to Bovine viral diarrhea virus, Bovine coronavirus, and Bovine herpesvirus 1. Immunoperoxidase-3,3′-diaminobenzidine (DAB). Bar = 17 μm.
Determination of immunoreactivity in other neoplasms that would be included in the differential diagnosis (e.g., oral spindle cell sarcomas). 46,48,50,53
Comparison of immunoreactivity between non-neoplastic and neoplastic tissues (antigen expression may be lost or expressed de novo in the neoplastic phenotype). 51
Determination of differences in immunoreactivity in tumors with variable cell phenotypes (e.g., well-differentiated and poorly differentiated) in the same tissue or different tissues (e.g., melanoma). 45,48
Determination of cross-reactivity of an antibody targeting a particular cell type in metastatic tumors that can be present in the same organ (e.g., hepatocellular tumors and tumors metastasizing to the liver). 49
Determination of differences in reactivity between native and mutated antigens (e.g., p 53).
Determination of prevalence of immunoreactivity of various antibodies in a variety of tumors or tissues to determine the relative utility of a particular antibody for supporting the diagnosis of a cell type based on the IHC reaction. 51,53
Species cross-reactivity. This problem is unique to veterinary medicine. Although some antigens can be detected under similar conditions in different species, it should be assumed that antigen detection will vary among species. 52 Identical antibody clones that target the same antigen can differ in reactivity among species (Fig. 10). Detecting an antigen in a new species may be difficult and laborious. Nevertheless, it is advisable to restandardize (optimize) a test for each species, including incubation times, concentration of the primary antibody, and AR. A literature search or consultation with the antibody manufacturer and extensive testing of similar cases in a given species are recommended to determine species cross-reactivity. For diagnostic purposes, an IHC report should state the degree of confidence in the results based on the laboratory's or others' experience (including published material). The use of tissue arrays from different animal species permits rapid screening for interspecies cross-reactivity. 31,39,40,43 Other advantages of the tissue microarray are savings in reagents and reduction in results variability; disadvantages include a) selection of the sample is critical because of its small size, and b) highly qualified personnel are needed to prepare the array.
Effects of fixation, postfixation treatments, and equipment on immunoreactivity
Different fixatives have different effects on immunoreactivity. For each fixative, full standardization of a test must be done, including incubation conditions, dilution of the primary antibody, and AR method (see “Standardization” section). Standardization is usually done on tissues with known fixation time (e.g., 1–2 days).
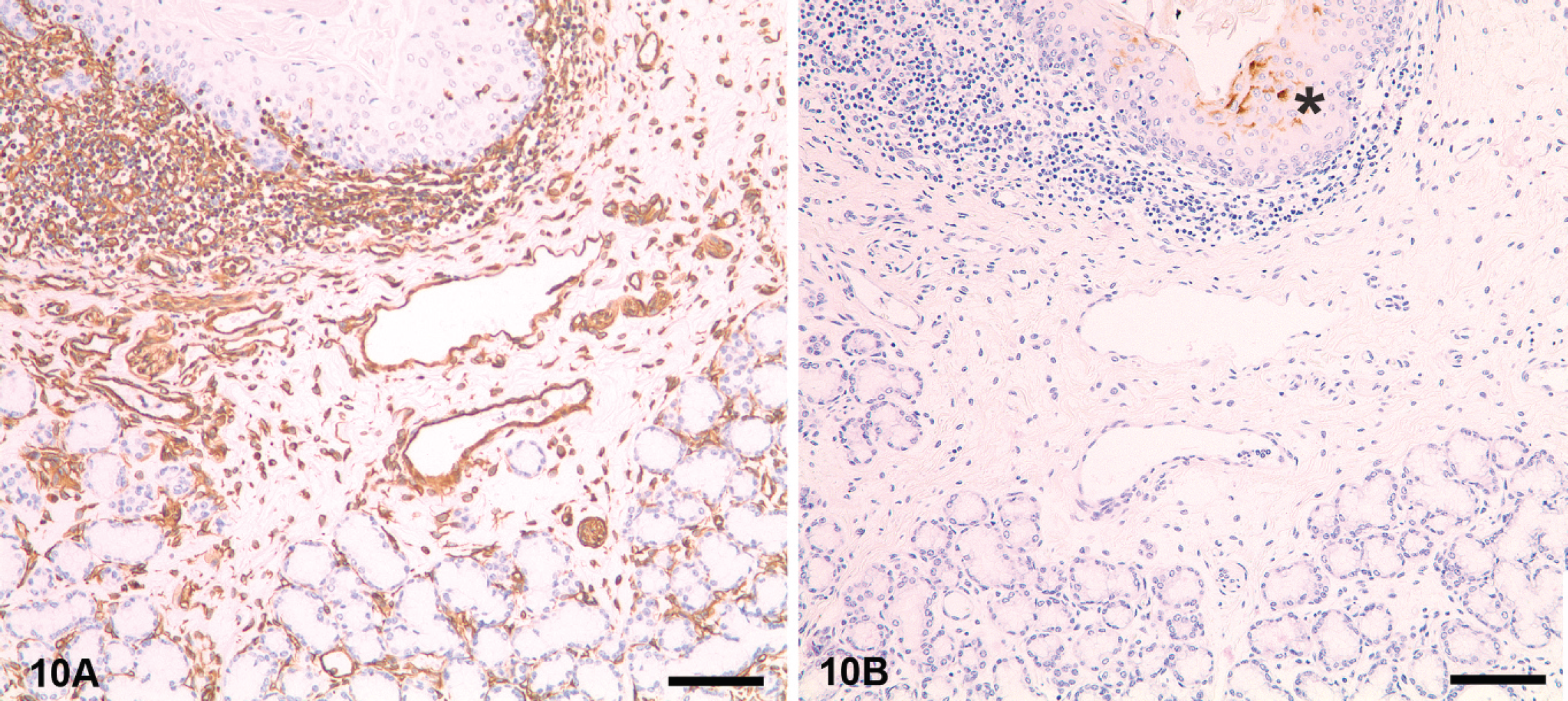
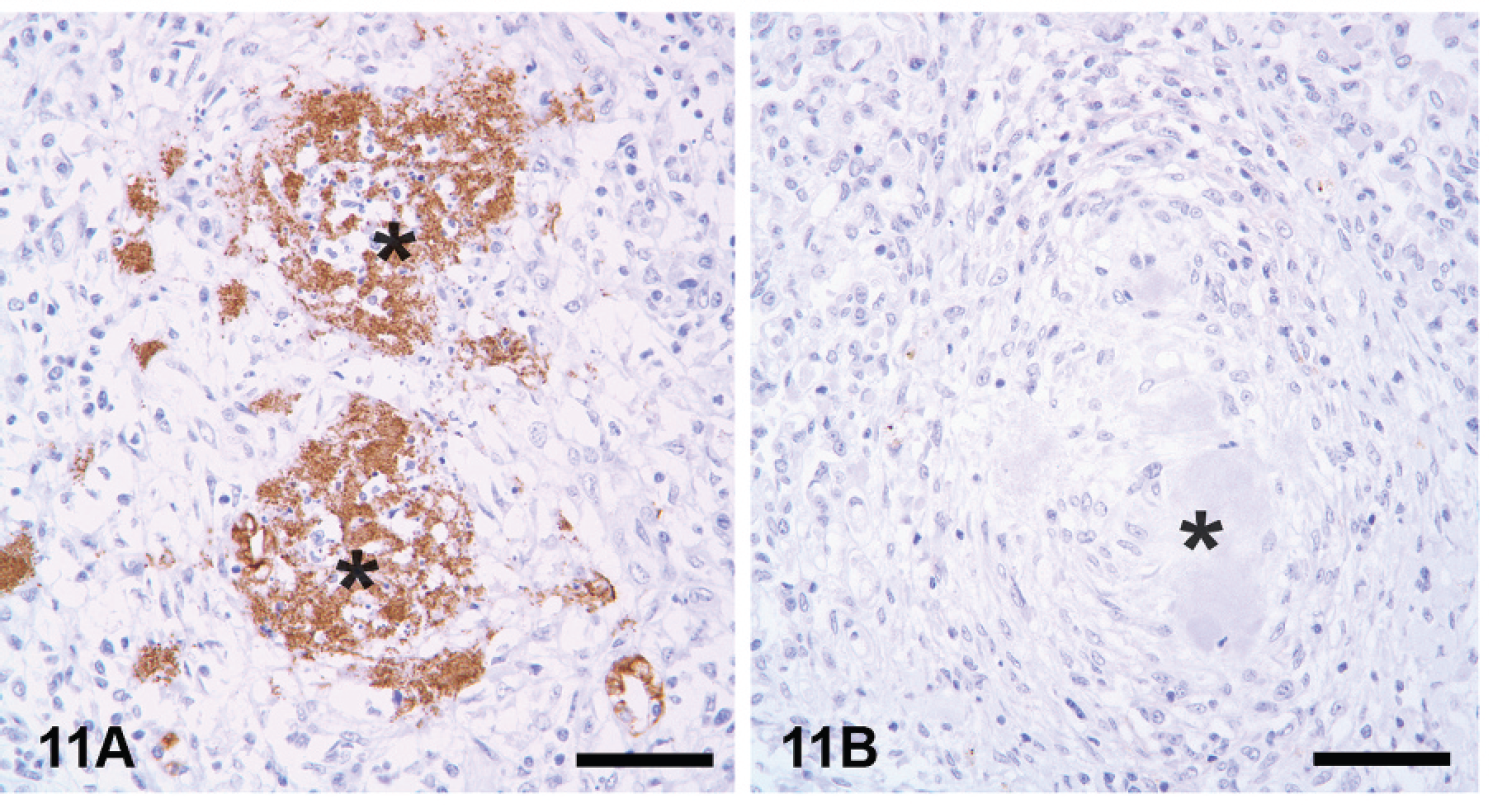
Fixation kinetics studies are recommended for each antigen or epitope. A positive tissue control should be fixed for different durations (e.g., 1, 2, 4, 7, 10, 14, 21, 20 days) and tested under identical conditions to determine the effects of fixation time on reactivity (i.e., intensity and number of cells or organisms detected; Fig. 11). 45,48
Automated stainers are designed to duplicate manual staining procedures and can be used to ensure uniform application of all steps of the process. Thus, the use of automated equipment offers a uniform and standardized microenvironment for testing, which will, in turn, result in intralaboratory run-to-run consistency. 64,77 When results among different laboratories differ considerably, these technical differences should be considered as a possible cause.
Storage of paraffin blocks and tissue sections
It is the authors' and others' opinion that paraffin blocks remain stable for years in terms of antigenicity. However, only controlled studies on the effect of nucleic acid detection on archival paraffin blocks have been published. 25 Any deleterious effects of prolonged storage on tissue antigenicity in paraffin blocks, if they happen, may differ among antigens. For practical purposes, consider this factor if inconsistent IHC results occur when using old paraffin blocks.
Storage of unstained paraffin control tissue sections increases efficiency but may adversely affect immunoreactivity (tissue section aging), depending on the antigen of interest. 24 Photo-oxidation of tissue sections is involved in loss of antigenicity. 13 Tissue section ageing is a rather common problem with nuclear antigens (e.g., Ki67, estrogen receptor, p53). 11,19,25,30,34,36,38,42,54,70,72 When there is decreased intensity or loss of reaction in stored control tissue sections, repeat the test with another stored control tissue slide and a freshly cut control tissue section. If the change is limited to stored sections, discard any remaining unstained control slides.
What constitutes a positive/significant result?
There is no single answer to this question. Immunohistochemical assays detect analytes (infectious agent or tumor marker antigens) in tissues but do not necessarily diagnose disease, which may require interpretation of IHC results in correlation with other clinical or laboratory findings. For an infectious agent, detecting one organism indicates infection, but it is another matter to prove that the agent caused disease. For neoplasms, this answer is even more difficult. In the literature, the percentage of positive cells required to confirm tissue origin of a tumor varies considerably, and often this striking variation is reported for detection of the same antigen by different authors. Some pathologists prefer the words “detected” or “not detected” rather than “positive” or “negative.” This seems semantic but underscores not only the need to interpret subjectively a colored reaction, but to do so in the context of the disease. 61
Controls
Positive tissue control
Positive tissue control is defined as tissue that is known to contain the antigen of interest detected by identical IHC methods to those used in diagnostic cases. Fresh-fixed surgical specimens (or biopsy tissue) are preferred over necropsy material whenever possible, since autolysis may affect staining results. Positive tissue controls must be fixed and stained in the same way as the diagnostic case tissue for every antibody and procedure used. 55,64 The presence of the antigen in the tissue control should be confirmed by another method (e.g., PCR, virus isolation). For most laboratories, control tissues are generally obtained “in-house” because variables such as fixation and processing are the same as those used with diagnostic case material. Control tissues obtained from another laboratory or from a commercial source may have been fixed/processed in a different way than the test tissue. However, the use of in-house standards does not ensure reproducibility of a particular IHC assay between different laboratories.
Multi-tissue blocks and cell lines. Various methods for the simultaneous study of multiple FFPE tissues in a single histologic section have been reported. The “sausage technique” has been used heavily in the past. 10 In this method, long thin strips of fixed tissues are drawn into a tube of unfixed small intestine. The entire sample is then fixed, resulting in a tight column that can be cut into blocks and embedded. Paraffin-embedded tissue microarrays have been proposed as a reference control standard for human IHC laboratories, primarily for use in tumor diagnosis. 29,39,40,43 It is recommended that validation studies be carried out on multi-tissue control blocks containing both known-positive and known-negative normal and tumor tissues. 64 In veterinary medicine, species-specific tissue microarrays are not commercially available. Species-specific tumor cell lines are being used for IHC controls (e.g., IHC for HER-2/neu). 55,57 This approach provides an identical tissue control among different laboratories and is an excellent way to compare results among them.
Reference control tissues for infectious diseases. The presence or absence of a particular infectious agent in a tissue should be assessed by previously published IHC reactivity patterns (if available) plus another non-IHC diagnostic method. 8 For example, Listeria monocytogenes–control tissue could be assessed by IHC (immunoreactivity of extracellular or phagocytized bacilli) and by bacterial culture. Similarly, Bovine viral diarrhea virus (BVDV)–control tissue could be assessed by IHC (immunoreactivity in cytoplasm of infected cells) and by virus isolation or PCR. In other instances, the presence of characteristic lesions for a particular disease (e.g., intranuclear inclusions observed with hematoxylin and eosin [HE] staining) can be used as a standard reference control.
Species reactivity differences. Eyelid, horse. This tissue was incubated with two different antibodies to vimentin, a mouse monoclonal antibody (
Effects of prolonged fixation.
Nonspecific staining. Knowledge of the expected antigen distribution of the reaction within a cell or tissue is mandatory before using a test for diagnostic purposes. In this case, an antibody to CD79a stained only the nucleus of plasmacytoma cells. This reaction is not considered specific based on the current knowledge of the location of this antigen (cytoplasmic and cytoplasmic membrane). Bar = 60 μm. The lower inset is a detail of the aberrant staining; the upper inset depicts a typical cytoplasmic staining of CD79a in a plasmacytoma. Immunoperoxidase-3,3′-diaminobenzidine (DAB). Both inset bars = 5 μm.

Equipment maintenance. One of the possible causes of unusual staining in immunohistochemistry is inadequate maintenance of equipment. The heat unit (arrows) of the steamer needs to be free of salts that will progressively deposit when using tap water.
Immunohistochemical protocols for infectious diseases should, whenever possible, also be species-matched with test specimens; alternatively, tissues from other species affected with the same infectious agent can be used (e.g., BVDV identified in diagnostic case material from cervids using a bovine control tissue). Nonspecific binding of primary antibody (species-specific molecular mimicry) or secondary (linker) reagents may occur among different species. A good example is the use of goat polyclonal antiserum specific for Neospora caninum on goat tissue. The anti-goat IgG secondary reagent used in the IHC assay can bind excessively to endogenous goat IgG in the tissue resulting in such severe background “noise” that visualization of individual immunoreactive tachyzoites is impossible.
Reference controls for neoplastic diseases. Many times, the only way to verify the presence of an antigen is by morphology, which is not always an accurate predictor for the presence or absence of a particular antigen. Furthermore, a negative result for tumor markers does not necessarily rule out a particular neoplasm because the cell marker of interest may not be expressed in a very poorly differentiated cell population. Ideally, controls for tumor cell markers should consist of the neoplastic tissue of interest, which expresses different intensities of reactivity. 64 Control tissues for IHC tumor markers should also be species-matched with test specimens because the detection of an antigen in a particular tumor and animal species does not guarantee similar results in a different animal species.
Negative tissue control
Negative tissue control is defined as tissue that is known not to contain the antigen of interest. 64 At least 1 ancillary test (e.g., PCR, virus isolation) performed on the tissues/organ systems of the same animal should be used to rule out the presence of the antigen of interest. Both positive and negative in-house tissue controls should be used in each test and should be processed in the same manner as the case material. Similarly, when using multitissue blocks for antibody validation studies or as tissue controls, the specimens must be fixed and processed in the same manner as the case material. 64 When dealing with cellular antigens (e.g., cytokeratin, vimentin), positive control tissue should have areas expressing variable amounts of the specific antigen. Weakly stained areas can be used to detect subtle changes in antibody sensitivity. 64 In practice, only 1 tissue control is used because of the common presence of both positive and negative cells within the control.
Internal positive tissue controls
Internal positive tissue controls are present in diagnostic case tissues. An example is the detection of smooth muscle markers or vimentin in normal blood vessels. The presence of positive staining in these areas indicates appropriate immunoreactivity. With this type of control, there is no fixation variable between the control tissue and the diagnostic case tissue. 14 Although some vimentin antibodies have been used to demonstrate overfixation effects due to their sensitivity to fixation, 55 this type of test may not be relevant with the current AR methods.
Reagent (antibody) controls
Negative reagent controls are used to confirm the specificity of the test and to assess the degree of nonspecific background staining present by omitting the primary antibody. Commonly, the primary antibody is replaced by 1 of the following methods: 1) antibody diluent, 2) same species nonimmune immunoglobulin of the same dilution and immunoglobulin concentration, 3) an irrelevant antibody, or 4) buffer. 55,64 These methods will assess the degree of cross-reactivity of the primary antibody, and the degree of nonspecific binding by the labeling (secondary) antibody and detection system; only methods 2 and 3 approach the true significance of negative controls. There are also commercially available ready-to-use universal negative reagents for rabbit and mouse primary antibodies.
If the diagnostic workup requires a panel of antibodies each of the same isotype and similar concentration and derived from the same species, the panel of antibodies itself may serve as a set of irrelevant reagent controls; thus, the need for multiple negative controls is eliminated. If this method is utilized, separate controls should still be run for each different type of protocol (e.g., for each protocol with a difference in the AR method or difference in the detection system). 64
Storage and handling of reagents
The shelf life of many reagents is directly linked to appropriate storage. The shelf life of many primary antibodies beyond that indicated by the manufacturer can be significantly extended with proper handling and storage. 6,67 However, use of a reagent beyond the manufacturer's expiration date is not covered by its warranty and has to be properly documented. General considerations for storage of reagents include:
Storage containers must be made of nonreactive material that does not alter the reagent by adsorption or polymerization of components within the reagent or addition of components to the reagent.
Plastics: Polypropylene and polycarbonate are nonreactive and are routinely used for storage of primary antibodies at −20°C or −70°C. Containers that can be tightly sealed are necessary to prevent desiccation. 18
Borosilicate glass: This material is used when storing reactive reagents. When storing diluted antibody preparations, addition of 0.1–1.0% bovine serum albumin (BSA) may be necessary to decrease polymerization and adsorption in any container used. 26
Primary antibodies should be stored frozen as concentrates in appropriately sized aliquots in keeping with manufacturer's recommendations. Freezing minimizes denaturation of proteins and reduces excessive mechanical action or contact with air. 26 Once thawed and diluted, the antibody preparation should be stored at 2–8°C. Repeated freezing and thawing of the concentrated antibody can result in significant loss of reactivity.
The majority of buffers are stored at 2–8°C to prevent microbial growth.
Sodium azide (NaN3) is often used in commercial concentrated primary antibody preparations as a preservative. For in-house antibodies, use 0.02% NaN3. To avoid enzyme inhibition by NaN3, use 0.01% merthiolate in conjugates. 26,66 Do not use preservatives in buffers or antibody diluents.
Close inspection of buffers and other reagents for signs of microbial contamination or precipitation of salts is necessary each time the reagent is used.
Proper labeling of stored reagents is essential. Each reagent label should include the name of reagent, date made or opened, expiration date, and name or initials of individual who made or opened the reagent.
Stock solutions often have a longer storage life compared to working solutions. Working solutions can be prepared daily for immediate use or stored for short term.
Substrates (or chromogens) should be freshly prepared for each run or as directed by the manufacturer.
Storage at −70°C does not provide significantly longer shelf life than storage at −20°C. Storage of reagents in frost-free freezers is not recommended because of their programmed fluctuations in temperature that contribute to increased polymerization and denaturation of many proteins.
Routine temperature checks of all storage areas and appropriate relevant documentation are recommended.
Quality assurance/quality control and interpretation of IHC results
Quality reflects on the procedural and technical aspects of the IHC test and is aimed to reduce and correct deficiencies in an analytical process. Each step of an IHC test is defined by quality control (QC)/quality assurance (QA) standards that have been previously addressed. Quality control issues, as they pertain to daily records and the standardization of new primary and secondary antibodies and controls, are discussed here.
Daily quality assurance/quality control
Each laboratory should establish standard operation procedures (SOPs) for their routine histology laboratory. This would include daily, weekly, and monthly schedules for cleaning, maintenance, and monitoring logs, along with a daily check of equipment such as oven temperature for drying slides, temperature of water baths, pH meter, etc. Buffers should be made using distilled deionized water (ddH2O) and their pH checked and adjusted, if necessary, before use. When enzymes are used for AR, they should be prepared shortly before use. There are also ready-to-use commercial enzyme solutions for AR. Note the storage conditions and expiration dates for any reagent.
A worksheet should list the stains requested and number of slides to be stained. The slides should be labeled accordingly and double-checked against the IHC request forms submitted by the pathologist.
Fresh working solutions of primary antibody should be prepared according to their storage requirements. The expiration dates of the antibody and the working dilution should be indicated on the tube. Xylene/xylene substitute and graded alcohols used for deparaffinization, rehydration, and dehydration should be changed before they lose significant strength. It is recommended to use AR solutions only once; however, each laboratory should establish their own protocol for reusing AR solutions.
When using an automatic stainer, a “Before Staining Checklist” should be observed.
The following are some important items to include on the list:
Check to ensure there is room for waste; empty the waste containers if necessary.
Check quantities of reagents for the run: ddH2O, buffers, IHC kit reagents, and chromogen.
Check to ensure correct programming is being used for the run.
Check slide labels as the stainer is loaded.
Check the quantity and placement of the IHC reagents.
Remove all caps from reagent tubes.
The maintenance schedule for the automated stainer should be followed and documented.
Primary antibodies. The date received, date aliquoted, date of dilution, and expiration date must be written on the appropriate containers. New lots of antibody should be tested and titrated before use. It is best to prepare only the quantity of reagents required in a given run. Aliquots should be made of stock primary antibody in volumes convenient for preparation of working dilutions.
Secondary reagents. Most reagents are commercially prepared and standardized by the manufacturer. Reagents should be stored per manufacturer's recommendations. Batch numbers should be recorded. Keep in mind that guarantee of a reagent only applies when the product is used as specified by the manufacturer.
Control tissues and stains. See “Controls” section.
Internal quality assurance/quality control
A bi-annual internal QA/QC check for the quality of IHC slide interpretation is recommended. In large diagnostic laboratories, randomly selected slides should be reviewed by other pathologists on a rotational basis. The slides should be interpreted by the pathologist and a written evaluation of the quality of the previous interpretation should be provided. Written comments on the technical aspects and the overall quality of the slides should be made.
External quality assurance/quality control
The goal for interlaboratory comparisons is to document variations in reactivity (e.g., positive vs. negative, intensity, number of positive cells) among different laboratories and to set minimum QA/QC standards to create optimal protocols that could be shared by all laboratories. 74 Interlaboratory standardization is difficult. Even in human medicine, only a handful of IHC tests (e.g., Hercep test) have been validated in such a way that different laboratories could perform tests with consistent results, and not without controversy. 12,27,58 Veterinary diagnosticians are still a long way from achieving interlaboratory standardization. As a previous analysis pointed out, “Whether we like it or not, the practice of anatomic pathology is to some extent subjective, although we all strive for as much objectivity and reproducibility as possible in our daily work.” 23
There are 2 main approaches to interlaboratory standardization:
Technical equivalency testing of an IHC test. Unstained slides are distributed among different laboratories for performance of the requested immunostains; results are compared among different laboratories or with those of the reference laboratory. This test determines the quality and interlaboratory consistency of the IHC testing.
Diagnostic equivalency testing using IHC testing as an adjunct. In this case, both the pathologist's proficiency and the quality of IHC testing are evaluated. A brief history and description of the tissue examined is included with tissue sections. The pathologist must make a diagnosis based on routine (e.g., HE) staining and support it with appropriate IHC testing. Results could be reviewed by an established committee that would address possible differences among participating laboratories. 73
The current working proposal of the Pathology Committee for the AAVLD Program for Interlaboratory Comparison of Infectious Agent Immunohistochemical Assays recommends having a reference laboratory (proposed to be the National Veterinary Laboratory Services, Pathobiology Section, Ames, IA) send participating laboratories a set of unstained FFPE sections for immunostaining and interpretation. Results should be qualitative (detected/positive, not detected/negative) and not quantitative. Participating laboratories would return the slides and interpretations to the reference laboratory for evaluation and feedback. For cellular antigens, a similar approach may be considered in the future.
Immunohistochemical report and interpretation
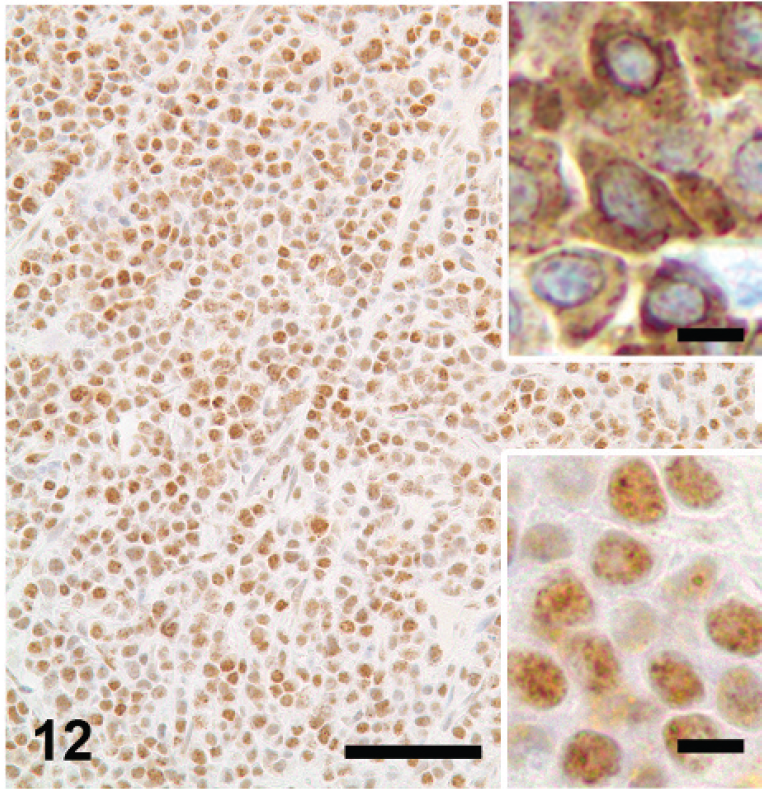
If not included in the standard report format, the IHC report should contain demographic information pertinent to the case, the tissue that was tested, and the antibody used, or the antigen in question should be listed. Other details of the IHC test should be filed in the laboratory. Results for infectious antigens have to be reported as “detected” or “not detected.” In addition, the interpretation for infectious disease IHC/immunocytochemistry (ICC) tests should include a disclaimer that any lack of detectable antigen may be a result of the absence of the antigen in this particular section and/or caused by technical aspects, such as prolonged fixation. In the case of tumor markers, it may be important to include a description of the cellular location (e.g., cytoplasmic, membrane, nuclear) and intensity of staining and percentage of positive cells, along with an interpretation of the significance of the test results (Fig. 12). When the antigen location is atypical, the report should mention this. Personnel who read IHC slides should be familiar with the antibodies used and their specificity and sensitivity. The authors highly recommend having a trained veterinary pathologist for QA/QC purposes review daily IHC staining.
Troubleshooting guide
Weak or no staining of positive control and weak or no staining of test slides
Consider the following:
Problem: all slides from some primary antibodies in the run are affected. Action: check for inadequacy of the primary antibody, method incompatibility, primary and link antibody incompatibility, or inadequate AR (Fig. 13).
Problem: the whole run is affected. Action: check assay log and adequacy of reagent volumes and sequence of reagent delivery to the slide. Determine if reagents were delivered to all slides (e.g., buffer, chromogen).
Problem: if it is hit-and-miss throughout the run, consider technical problems or problems with the tissues. Action: check for inadequate sequence of reagents, unbalanced autostainer, and inadequate drop zone.
Checklist assuming that the positive control tissue has been proven:
Review assay logs/datasheets/checklists to be sure that no step was omitted and that all steps were done in correct sequence.
Review assay logs/datasheets/checklists for appropriate incubation times and temperatures.
Check protocol for appropriate pretreatment or AR.
Make sure the linking antibody is compatible with the primary antibody.
Check for out-of-date reagents or antibodies and ensure that they have been properly stored according to package inserts.
Ensure that the primary antibody was properly diluted.
If a reagent was stored frozen, determine that it was not repeatedly frozen and thawed.
Check deparaffinization protocols and reagent quality (e.g., not overused).
Ensure that the substrate-chromogen was freshly and properly prepared.
Determine that the slides did not dry out at any time during the procedure.
Check the pH of the buffer solutions.
Other less common but possible problems:
Sodium azide in the wash buffers may inhibit peroxidase reactions.
Certain commercial phosphate buffer solutions may inhibit alkaline phosphatase activity.
Use of inappropriate counterstain or dehydration steps with alcohol-soluble chromogens.
Exposure of tissues to temperatures of >60°C during embedding or drying.
Leaving too much buffer on slides after washes. This dilutes reagents.
Excessive counterstaining interfering with interpretation.
Prolonged storage of tissue sections.
Try a new positive control tissue.
Inadequate or no staining of test slide and adequate staining of positive control slide
Problem: this is a true negative result; the antigen in question is not present in the test tissue. Action: none.
Problem: the antigen is present in the test tissue, but in a concentration below the method's detection limits. Action: consider using an amplification procedure, or increasing the primary antibody concentration, incubation time, or temperature or a combination thereof.
Problem: the test tissue is over- or underfixed. Action: modify AR protocol.
Problem: the test tissue is from a different species than the control tissue and has different reactivity with the primary antibody. Action: validate the IHC test with same species control and test tissues.
The following are other potential but uncommon problems:
Excessively high (>60°C) embedding or drying temperature was applied to the test slide only (assuming the positive control tissue and the test tissue are on different slides). Action: check oven temperature.
Prozone effect occurred due to high concentration of the primary antibody in the test tissue. Action: retitrate the primary antibody for the test slide.
A technical error (e.g., inadequate determination of the drop zone by the autostainer; unbalanced platform of autostainer or slide rack(s); insufficient amount of reagents) has occurred. This may occur on the test slide but not the positive control slide (assuming the positive control tissue and the test tissue are on different slides) or the test tissue but not the positive control tissue (assuming the test tissue and positive control tissue are on the same slide). Action: check autostainer balance status and dropping zone, respectively.
No staining of positive control slide and adequate staining of test slide
Problem: technical error in the staining or handling of the positive control slide. Action: the log or checklist should be reviewed; if everything is in order, the assay should be repeated. For control blocks for infectious diseases, the antigen to be tested may not be present through the entire block.
Problem: tissue section aging. Lack of staining in the tissue section control might be a result of tissue section aging. Action: compare staining of stored tissue sections with a fresh cut control section.
Excessive background staining
Prestaining problems.
Inadequate fixation, necrosis, and autolysis
Tissue sections allowed to dry out. Action: reduce incubation time; incubate in a humidified chamber.
Sections not completely deparaffinized. Action: use fresh dewaxing solutions.
Slide adhesive inappropriate or too thick. Action: use adhesives specific for IHC or positive-charged slides.
Tissue section too thick. Action: prepare thinner sections.
Inappropriate AR used. Action: re-evaluate AR conditions.
Incubation temperature too high. Action: reduce temperature.
Blocking problems.
Endogenous enzyme activity not suppressed. Action: increase concentration of blocking agent.
Inadequate protein blocking. Action: use a different blocking agent.
Inadequate blocking of endogenous avidin-binding activity. Action: use an avidin–biotin blocking step or use a nonavidin–biotin detection method (Fig. 14).
Inadequate blocking of endogenous biotin. Action: use an avidin-biotin blocking step or a nonavidin–biotin detection method.
Blocking serum from improper species. Action: use blocking serum from same species as the link (secondary) antibody.
Primary antibody problems.
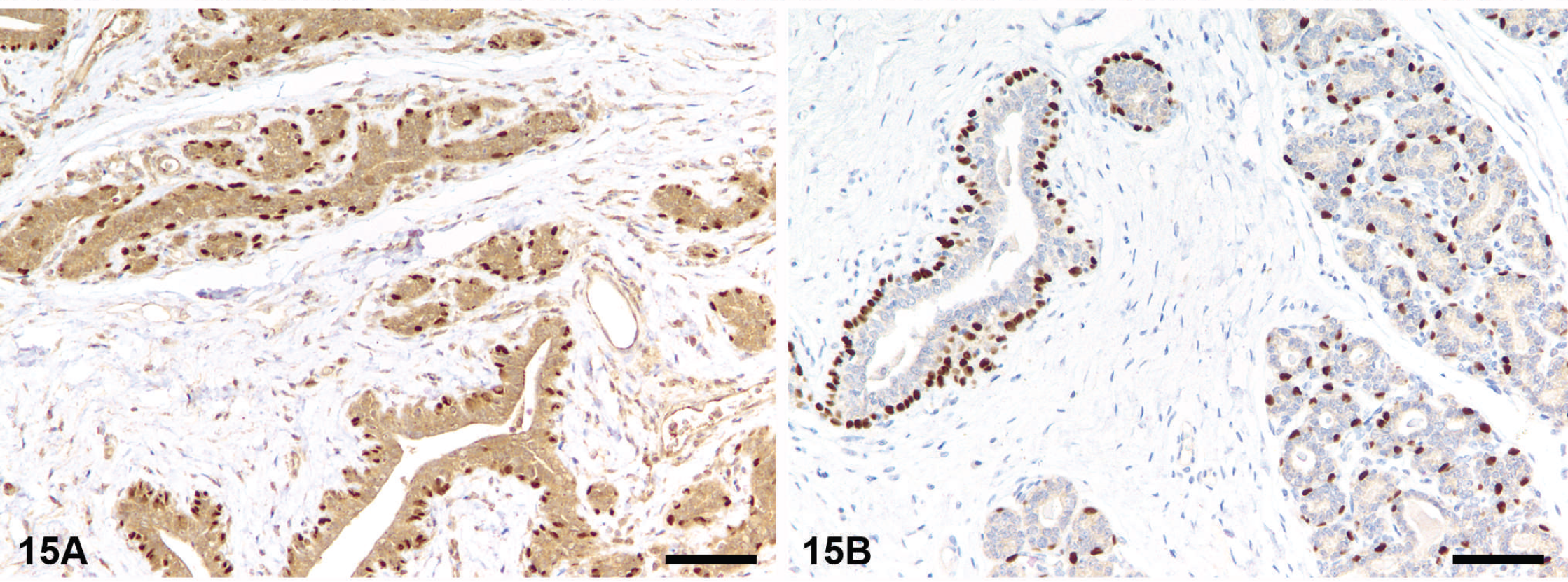
Primary antibody too concentrated. Action: retitrate primary antibody (Fig. 15).
Primary antibody incubation time too long. Action: reduce incubation time.
Primary antibody is from a similar or identical species as the test tissue (e.g., mouse on mouse [MOM], rat on mouse). Action: use specific protocols (i.e., MOM or similar kits) or additional blocking steps.
Inadequate buffer washes (inappropriate buffer ion concentration). Action: modify ionic strength of the buffer solution.
Secondary antibody problems.
Secondary antibody and label concentration too high.
Secondary antibody and label incubation time too long.
Buffer washes insufficient.
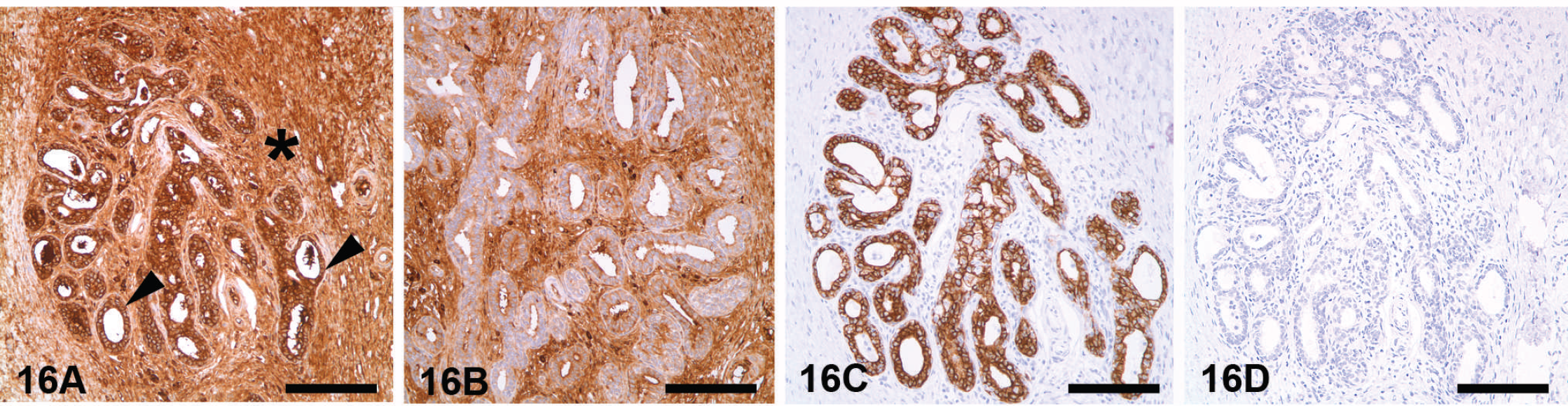
Secondary antibody recognizes endogenous (tissue) immunoglobulins (Fig. 16).
Microbial contamination of primary antibody solution. Action: reformulate from fresh stock. Add antimicrobial preservatives. Respect proper storage conditions for antibody solutions.
Chromogen and counterstain problems.
Chromogen concentration too high. Action: reduce concentration of chromogen.
Chromogen allowed to react too long. Action: reduce incubation time with chromogen.
Buffer washes insufficient. Action: prolong buffer washes.
Background caused by endogenous avidin–biotin activity (EABA). Kidney, dog. Background staining caused by antibody concentration. Mammary gland, dog. Background staining caused by secondary (link) antibody. This is a common problem when using secondary antibodies recognizing antibodies from the same species as the tissue examined. In this case, 2 detection systems (LSAB+ for A and B) and EnVision+ (for C and D) were used. Goat tissue stained for cytokeratins using LSAB+ detection system (the secondary antibody recognizes endogenous goat immunoglobulins) has a strong nonspecific staining in both the positive (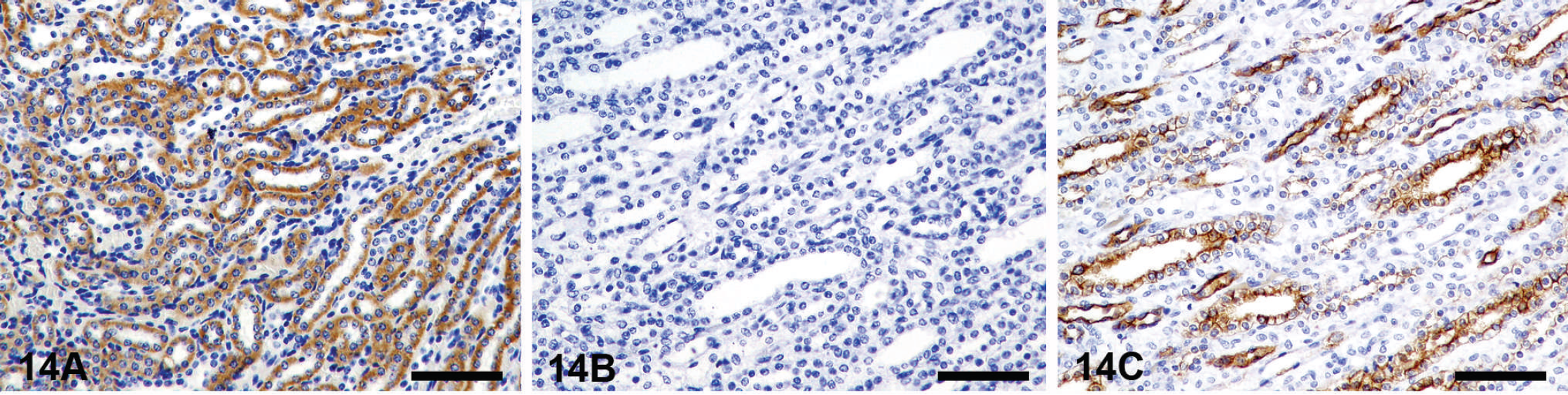
Counterstain obscures the IHC reaction. Action: use a different counterstain that does not interfere with IHC staining.
An excellent review of troubleshooting is available by Atwood in Immunohistochemical Staining Methods. 5
Conclusion
Immunohistochemistry is a well-established ancillary technique to facilitate the diagnosis of infectious and neoplastic processes in animals. This article has reviewed the main factors involved in the IHC test, from the preservation of samples and preparatory steps, followed by the immunohistochemical reaction and visualization of the reaction, to the interpretation and generation of an IHC report. Each step of the IHC test may require troubleshooting and a guide for this is also included. One goal of this article is to suggest guidelines to help veterinary laboratories establish standardization and validation procedures in diagnostic IHC, thus providing a method of quality control and quality assurance in a very subjective discipline.
Acknowledgements
The authors are members of the AAVLD Subcommittee on Standardization of Immunohistochemistry.