
Abstract
Clinical signs of prion disease are not specific and include a variety of differential diagnoses. Serological tests and nucleic acid-based detection methods are not applicable to prion-disease-agent detection because of the unusual nature of the infectious agent. Prion-disease diagnosis is primarily conducted by means of immunodetection of the infectious agent, typically by at least 2 distinct procedures with immunohisto-chemistry and Western blot being the most informative. These approaches differ in the need for formalin-fixed and frozen or fresh tissue respectively. This work describes a method for the detection of the disease-associated isoform of the prion protein by Western blot using formalin-fixed tissues. The approach requires only minimal modification of existing Western-blot procedures and could readily be incorporated into existing detection schemes for confirmatory purposes when fresh or frozen tissues are unavailable.
Keywords
The normal cellular isoform of the prion protein (PrP C ) is a host encoded protein that can undergo a conformational change to a disease associated form termed PrPSc. PrPSc is the causative agent of a class of fatal transmissible neurodegenerative diseases, the transmissible spongiform encephalopathies (TSEs). 9 Clinical signs of TSEs are nonspecific and microscopic lesions of spongiform encephalopathy may not be present in all prion diseases (e.g., cross species transmission of TSEs). 1 Specific immune responses have not been detected in TSE-infected organisms, and serological tests to obtain evidence for the presence of PrPSc are not available. Most notably, nucleic acid-based detection methods are not applicable because of the fact that the infectious agent is a protein.
Diagnosis of a TSE today is primarly conducted by means of immunodetection, including immunohistochemistry (IHC), Western blotting, and enzyme-linked immunosorbent assay (ELISA)-based approaches. 4,5,10 Infected tissues contain both PrP C and PrPSc, and most PrP-specific antibodies react with both PrP isoforms. For Western blotting and ELISA, this is overcome by limited proteolysis, typically using proteinase K, such that the protease sensitive PrP C is removed leaving only the relatively protease resistant PrPSc. IHC does not depend upon proteinase K for removal of PrP C , however, it does require significant training and expertise for reliable differentiation of positive and negative cases. 2,7 Each approach has advantages and disadvantages, and it is preferred that more than 1 technique (typically both IHC and Western blot) would be applied to a given case before a final diagnosis is made. 4 Confounding the combined use of IHC and Western blot for a complete diagnosis of a TSE is the fact that Western-blot approaches require either fresh or frozen tissue whereas optimal conditions for IHC are formalin-fixed tissue that has never been frozen. Thus, appropriate sample handling can be problematic requiring refrigeration without freezing or preservation of 2 independent samples. Although having both fixed and fresh or frozen tissue will undoubtedly still be ideal, the ability to use formalin-fixed tissue for Western-blot confirmation of IHC results could alleviate some of the sample handling issues and may prove useful for collection of field cases of TSE-suspect animals.
Methods for protein extraction from formalin-fixed tissues for the purposes of Western-blot analysis have been presented in the literature, but to date, none of these has proven useful for PrPSc detection. 3,11,14 An approach for detection of PrPSc in formalin-fixed tissues is presented here that requires only minimal adaptation of existing Western-blot procedures and could be incorporated into any existing Western-blot protocol.
Sheep brainstem tissue was harvested from animals intracranially inoculated with scrapie or negative control animals all housed and cared for under approved National Animal Disease Center animal care and use committee approved protocols. Collection took place during the postmortem examination and tissues were placed immediately into 10% buffered formalin for a minimum of 2 weeks and up to 6 years. Experimental details for all samples depicted in this work are provided in Table 1. Archived material totaling 10 samples with formalin storage times ≥2 years were analyzed as well (data not shown).
Tissue preparation was by an adaptation of a method developed by Miller et al. for purposes of PCR-based detection of bacterial pathogens. 6 Tissue (∼500 mg) was removed from the 10% buffered-formalin solution and placed in 95% ethanol for 48 hours to remove the formalin. The tissues were then placed in PBS pH 7.5 for 48 hours to remove the ethanol. At this point, tissues were either frozen at −20°C for later use or extracted for immediate use. Tissue was homogenized at a final concentration of 20% (w/v) in 50 mM Tris, 0.5% Tween 20 pH 7.5. The homogenate was boiled for 10 minutes and then rapidly frozen in a dry ice, ethanol slurry. This cycle was repeated 5 additional times. Samples were analyzed at this point or digested with proteinase K. a Those samples that were digested with proteinase K were digested using a final enzyme concentration of 4 U/ml (80 μg/ml) at 37°C for 1 hour. The digestion was stopped by addition of Pefabloc b to a final concentration of 0.1 mg/ml. Nonfixed, frozen tissues were homogenized as indicated for the fixed samples and then analyzed by sodium dodecyl sulfate-polyacryl-amide gel electrophoresis (SDS-PAGE) and Western blot or digested with proteinase K, as indicated above, before analysis. Nonfixed samples were not subjected to the freeze-boil cycles.
Sample status and time in formalin.
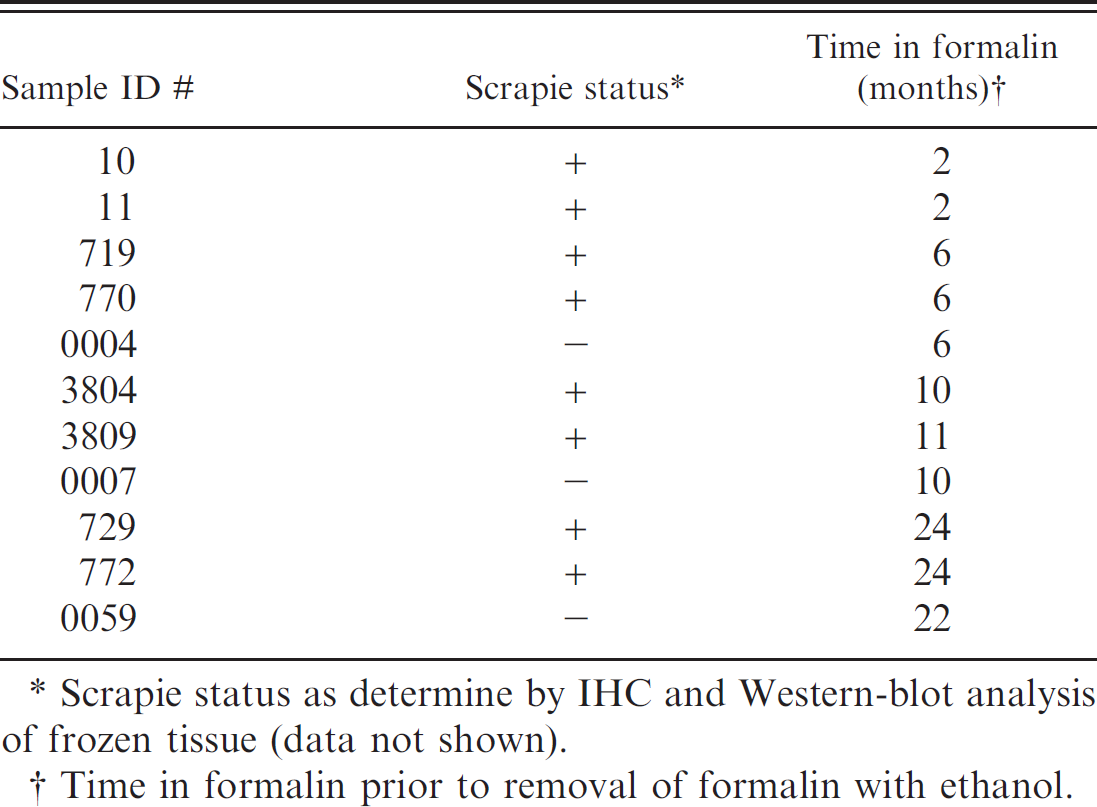
Scrapie status as determine by IHC and Western-blot analysis of frozen tissue (data not shown).
Time in formalin prior to removal of formalin with ethanol.
Samples were diluted 1:2 in 2− SDS-PAGE sample buffer such that between 1 mg and 4 mg tissue equivalent of tissue sample was analyzed by standard Western blotting procedures. The tissue equivalent for loading was empirically derived with the goal of determining a range of loading that would provide a reasonable signal for each case while not requiring any concentration or enrichment steps. A tissue equivalent of 1 mg represents a reasonable starting point for analysis whereas 4 mg represents approximately the maximum amount of tissue that can be loaded without concentration or enrichment. For Western blot, a 4%-20% commercially prepared SDS-PAGE gel c was loaded and run according to the manufacturer's instructions, blotted to a polyvinylidnene difluoride (PVDF) membrane and blocked with 3% bovine serum albumin. Western-blot detection was conducted using mouse anti-PrP monoclonal antibodies P4 d or 6H4 e at a 1:10,000 dilution (0.1 μg/ml) or F99/97.6.1 f at 1:1,000 dilution (1 μg/ml) as the primary antibody. A biotinylated sheep anti-mouse secondary antibody g at 0.05 μg/ml and a streptavidin-HRP conjugate, g were used in conjunction with the ECL Plus detection system g and visualized on either a Kodak In vivo F h or a Typhoon i imaging system. Primary antibody incubations were conducted with the membrane at either room temperature for 1 hour or 4°C overnight (≥12 hours). Secondary antibody and streptavidin-HRP conjugate incubations were conducted at room temperature for 1 hour.
Using the method described here, material extracted from formalin-fixed tissue for analysis by Western blot accurately defined the scrapie-positive or negative status of all samples that were treated with formalin for 2 years or less. The scrapie status as determined by IHC and Western blot (data not shown) of each sample and the time in formalin before analysis are shown in Table 1.
Western-blot analysis of representative frozen tissues are shown in Fig. 1A. The 3 bands characteristic of un-, mono-, and di-glycosylated PrPSc between 20 and 30 kD were clearly present in all positive samples. 13 As expected, proteinase K digested all detectable PrP C under conditions of the experiment and the lane containing a negative sample was observed as an empty lane.
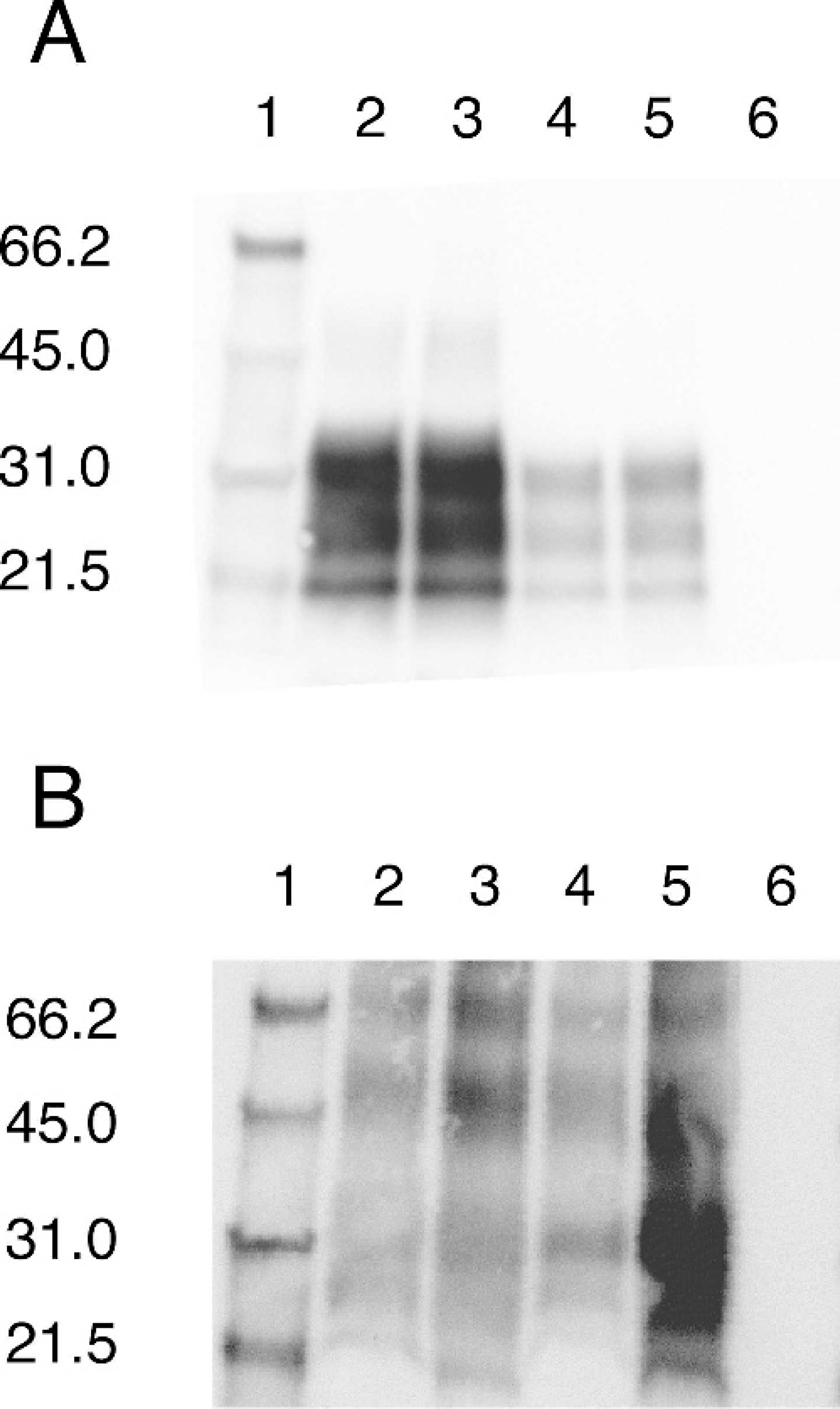
Representative Western blot of frozen
Shown in Fig. 1B are formalin-fixed and extracted samples that had been preserved in formalin for up to 2 months. Prion proteinSc can be detected for up to 2 years (Fig. 2). Scrapie negative samples exhibited no immunore-activity. This occurs not only with monoclonal antibody P4 but with antibodies 6H4 and F99/97.6.1 as well (data not shown). Notable in the scrapie-positive, fixed samples in Fig. 1B and 2 are higher molecular weight species not present in the corresponding frozen samples (Fig. 1A and unshown data). These higher molecular weight bands are only present in the fixed, positive samples, and these bands are, in some cases, of higher intensity than the lower molecular weight monomeric species. A negative control blot corresponding to Fig. 1B, processed without the primary antibody, was run to confirm that the bands observed are specific to PrP and not an artifact of nonspecific interactions with either the secondary antibody or the detection system. No bands are observed for any samples (data not shown), thus the observed bands in the scrapie-positive lanes are specific to PrP and do not reflect nonspecific interactions with either the secondary antibody or the detection system.
In Fig. 2, the effects of proteinase K digestion on formalin-fixed samples are depicted for samples that have been preserved in formalin from 2 months to 2 years (see Table 1). Each digested sample is adjacent to the corresponding nondigested sample. Proteinase K digestion enhances signal intensity for the positive samples. As indicated previously, the negative control samples show no immunoreactivity regardless of whether or not proteinase K digestion was included.
These preliminary results from a limited number of experimental sheep scrapie cases demonstrate that Western blot may be a suitable detection system for PrPSc in formalin-fixed tissues. The approach presented here proved successful at accurately identifying both positive and negative sheep scrapie samples for all analyzed tissues subjected to up to 2 years of formalin fixation. By 6 years time in formalin, all PrP signal had been lost using the present approach (data not shown). The precise timing of this change was not established, only that it had not occurred at 2 years and was complete by 6 years. Such variability supports the need for standardization of formalin fixation for diagnostic and archival purposes. 12
As indicated previously, higher molecular weight species are present in scrapie-positive, formalin-fixed samples that are not present in either scrapie-positive, nonfixed samples or in scrapie-negative, fixed samples. Based upon this we conclude that these bands correspond to PrPSc oligomers formed by formalin crosslinks within the scrapie-associated fibrils. Because these bands may equal or even exceed the intensity of the bands at the anticipated molecular weight for PrPSc monomers and they are not observed in negative samples, any descrete band is consistent with that sample being a scrapie-positive sample regardless of the molecular weight. This may not hold true for all TSEs in all species and caution is urged in considering a sample positive unless the 3 characteristic bands in the ∼20–30 kD molecular weight range typically associated with PrP are present.
Anecdotally, it was observed that a relatively larger sample loading may be required for formalin-fixed tissues relative to frozen tissues (data not shown). Undoubtedly some of this arises because of the higher number of oligomeric species present. Prion proteinSc is present as at least 6 distinct bands in fixed tissues, many of which exceed 30 kD. This is in contrast to Western-blot analysis of nonfixed samples where 3 characteristic bands corresponding to partially digested un-, mono-, and di-glycosylated PrpSc are observed. 13 The precise molecular weight of these bands depends upon both the species and the TSE in question but are typically in the range from ∼20 to 30 kD. 8 Thus, with PrPSc being present in a larger number of species some of which include more than 1 PrP monomer, it is within reason that roughly twice as much material is required to obtain equivalently intense bands.
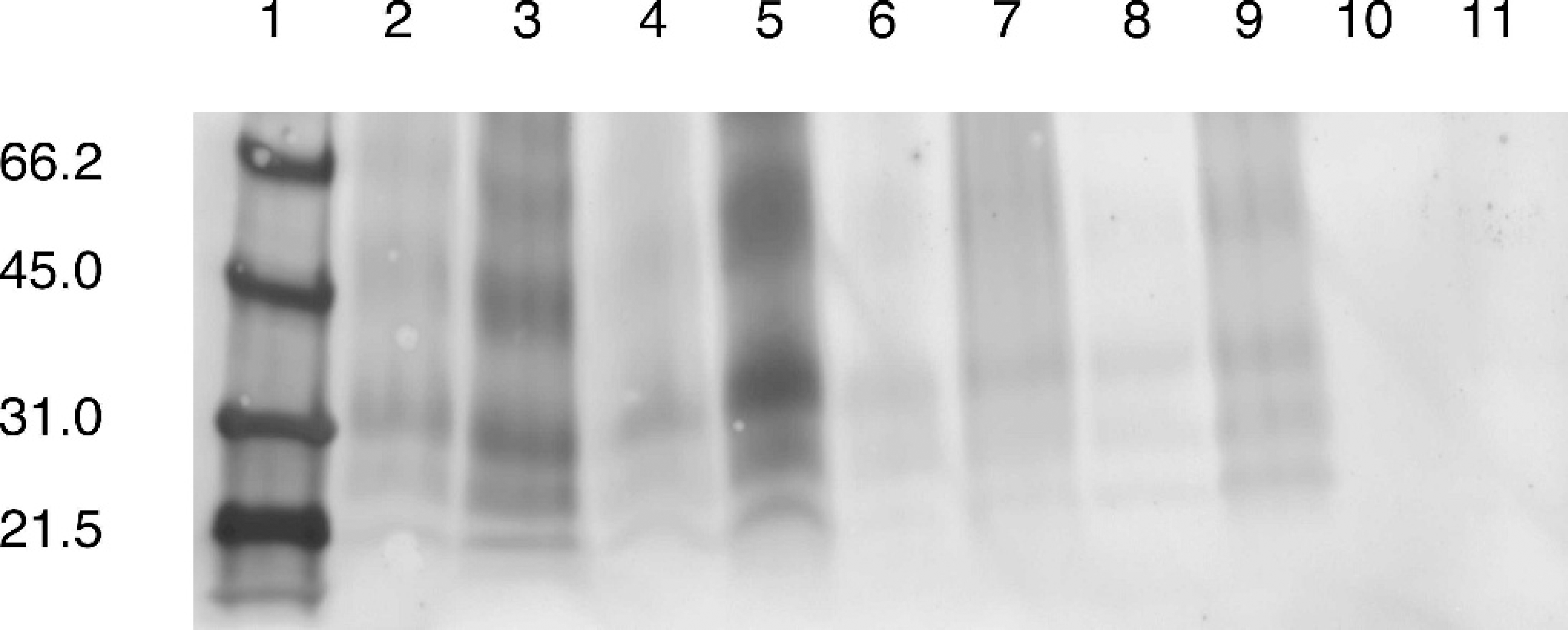
Western-blot analysis of proteinase
The results presented here indicate that PrPSc detection using Western-blot analysis of formalin-fixed tissues can be accomplished with only minimal modification of current Western-blot protocols. One notable difference is that PrP C is not observed in Western blots of formalin-fixed tissues, even when the standard method for removing this isoform, proteinase K digestion, is not included. This most likely occurs because of chemical modification of PrP C preventing detection with the tested monoclonal antibodies. Because of the absence of any detection of PrP C , the internal, positive control of an undigested sample typically afforded an investigator when using Western-blot detection of PrPSc is lost. However, the loss of the internal positive control is not a major limitation and it can simply be overcome by including an external positive control for the blotting procedure. Based upon this limited and preliminary study of sheep scrapie, positive immunoreactivity on a Western blot appears to be definitively positive with no discernable detection of PrP C . Use of proteinase K is still desirable because of the improved blot quality (Fig. 2), most likely as a result of improved tissue disruption, but based upon the samples analyzed here it does not appear to be an absolute requirement for the purposes of removing PrP C . Inclusion of the proteinase K digestion is encouraged because of the improved blot quality and the fact that it would prevent false positive cases in the event that PrP C signal on a Western blot is not precluded by the formalin fixation step.
Ultimately, this approach may be incorporated into a diagnostic regimen as a confirmatory technique eliminating the current necessity of having both frozen and formalin-fixed tissue. Frozen tissues should still be considered the preferred substrate for Western-blot analysis, however, in the absence of such tissues formalin-fixed tissue can be a suitable substitute.
Acknowledgements. We thank Dr. Mark Hall (Patho-biology Laboratory-NVSL, USDA, APHIS) for helpful discussion, Drs. Tracy Nicholson and Marcus Kehrli for critical evaluation of the manuscript, and Ms. Martha Church for technical assistance. Disclaimer: Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
a.
USB, Cleveland, OH.
b.
Roche Diagnostics, Indianapolis, IN.
c.
Pierce, Rockford, IL.
d.
R-Biopharm AG, Southmarshall, MI.
e.
Prionics, Schlieren, Switzerland.
f.
VMRD Inc, Pullman, WA.
g.
GE Healthcare, Piscataway, NJ
h.
Kodak, Rochester, NY.
i.
GE Healthcare, Piscataway, NJ.