
Abstract
Canine parvovirus (CPV) type 2 (CPV-2) emerged around 1978 as a major pathogen of dogs worldwide. In the mid-1980s, the original CPV-2 had evolved and was completely replaced by 2 variants, CPV-2a and CPV-2b. In 2000, a new variant of CPV (named CPV-2c) was detected in Italy and now cocirculates with types 2a and 2b in that country. The CPV-2c has also been reported from single outbreaks in Vietnam and Spain. This study was conducted to determine if CPV-2c occurs in the United States. Thirty-three fecal samples were collected from dogs in 16 states between April 2006 and April 2007 and were tested for CPV using real-time polymerase chain reaction (PCR). Positive samples were further tested using conventional PCR and minor-groove binding TaqMan PCR assays to determine the viral type and to differentiate vaccine strains from field strains. Twenty-seven samples were positive for CPV, 7 of which were CPV-2c from 5 states: Arizona, California, Georgia, Oklahoma, and Texas. Of the 7 isolates, 4 differed from European CPV-2c isolates by 2 additional single-nucleotide mutations at positions 4076 and 4104, the latter of which produces a ThrAla change at residue 440 located near a major antigenic site. The coast-to-coast geographic distribution of the states in which CPV-2c was detected strongly suggests that this new CPV variant is probably widespread in the United States. The continuous evolution of CPV requires that monoclonal antibody-based and nucleic acid-based diagnostic assays should be periodically checked for sensitivity on prevalent CPV strains.
Canine parvovirus (CPV) emerged in the late 1970s, presumably from feline panleukopenia virus or from a wild carnivore parvovirus via genetic mutations and evolution, and it rapidly established itself as a major viral pathogen of dog populations worldwide. 1,3,12 The new highly pathogenic virus was named CPV type 2 (CPV-2) to distinguish it from the less pathogenic and antigenically unrelated CPV type 1 or minute virus of canines. 4 Shortly after its emergence, the original CPV-2 continued to evolve and was completely replaced by 2 variants, CPV-2a and CPV-2b, by the mid-1980s. 17,19 In the past 20 years, CPV-2a and CPV-2b have cocirculated in various proportions among dog populations worldwide. 22
In 2000, a new variant of CPV (subsequently named CPV-2c) was detected in Italy 2 and is now widely distributed and cocirculating with types 2a and 2b in that country. 14,15 The CPV-2c has also been reported from single outbreaks in Spain 8 and Vietnam. 16 This new variant is distinguishable from CPV-2a/2b by the substitution of Glu in lieu of Asn or Asp at residue 426 of the capsid protein VP2; therefore, it is also referred to as Glu-426. This substitution involves a major antigenic site (epitope A) located on the 3-fold spike of the capsid protein 21 and results in a change in antigenicity that has made it possible to differentiate CVP-2c from CPV-2a/2b using monoclonal antibodies. 16
Genetic variation in a virus can adversely affect nucleic acid-based diagnostic assays if a change occurs in the primer binding region. Monoclonal antibody-based assays may also be adversely affected if genetic variation results in a change in the antibody binding site. Similarly, antigenic variation may negatively affect vaccine efficacy if biologically significant changes occur in major antigenic sites. For these reasons, it is important to continuously monitor the occurrence of novel genetic and antigenic types in viruses, such as CPV, that appear to continuously evolve. The objective of this work was to determine if the CPV-2c that has recently been described in Europe and Asia also occurs in the United States.
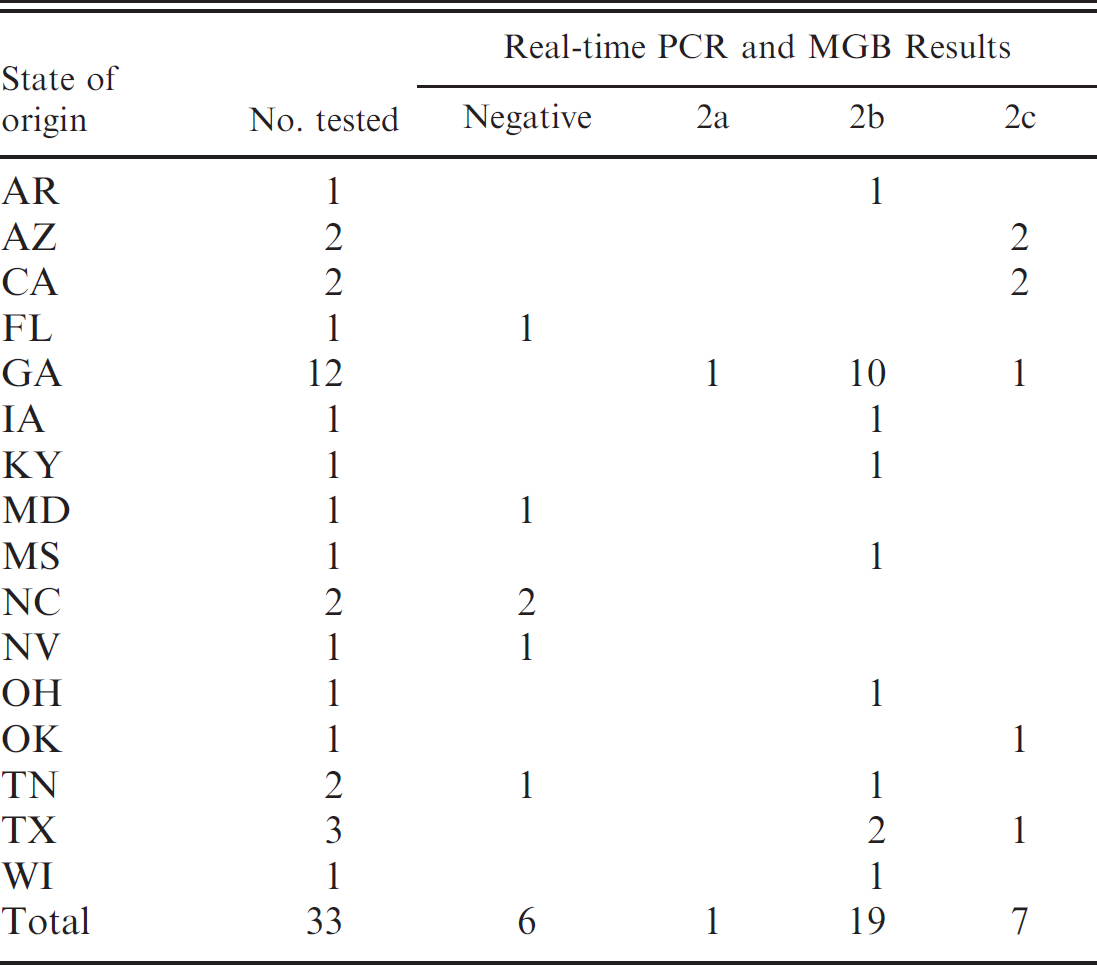
A total of 33 fecal samples from vaccinated (n=31) or unvaccinated (n=2) dogs were obtained by Merial Limited, to support epidemiologic surveying. The samples were collected between April 2006 and April 2007, and they originated from 16 states (Table 1). All samples were tested for CPV DNA using a previously described CPV-2 group-specific TaqMan-based real-time polymerase chain reaction (PCR) assay. 6 Samples that were positive using real-time PCR were further tested using minor groove binder (MGB) Taqman PCR assays. 5,7,9 and a conventional PCR to determine the viral type. The 33 samples were also used for virus isolation.
Template DNA was extracted from 10% (wt/vol) suspensions of fecal samples in Dulbecco's minimal essential medium (DMEM) using a commercial DNA fecal extraction kit. a Extracted DNA samples were stored at −20°C until tested. For each PCR reaction, DNA from previously typed CPV field isolates representing subtypes 2a, 2b, and 2c were included as positive controls, while a sample extracted from DMEM using the same
DNA fecal extraction kit was included as a negative control.
Results of real-time PCR testing and subtyping of CPV isolates.
The TaqMan and MGB probe assays were carried out in a 25-μl reaction containing 10 μl of template DNA or control DNA (both in duplicates), 12.5 μl of IQ Supermix, b 600 nM (TaqMan assay) or 900 nM (MGB probe assays) of primers, and 200 nM of probes. The thermal cycling parameters were as follows: activation of iTaq DNA polymerase at 95 °C for 10 minutes, 45 cycles of denaturation at 95°C for 30 seconds, and primer annealing/extension at 60°C for 1 minute. All reactions were conducted in an i-Cycler iQ Real-Time Detection System, c and the data were analyzed using the software included (version 3.0).
To determine if CPV subtype 2c isolates from the United States are similar to those described in Europe, a 583-segment of the VP2 gene generated using conventional PCR was sequenced. This segment includes the sequence of the amino acid residue at position 426 that was the basis for CPV-2c being classified as a different subtype than CPV-2a and CPV-2b. Conventional PCR was performed as previously described, 2,10 with minor modifications. Briefly, the reaction was carried out in a volume of 50 μl containing: 30 μl of Amplitaq Gold Master Mix (0.05 U/μl AmpliTaq Gold DNA polymerase, GeneAmp PCR Gold Buffer [30 mM Tris/HCl, pH 8.05, 100mM KC1], 480 μM dNTP, 5 μM MgCl2, and stabilizers), d 5 μl of deionized water, 5 μl (1 μM) of forward primer, d 5 μl (5 μM) of reverse primer, e and 5 μl of template DNA. The thermal cycling conditions were as follows: activation of AmpliTaq Gold polymerase at 10 minutes at 94°C; 35 cycles of denaturation at 94°C for 30 seconds, primer annealing at 50°C for 1 minute, extension at 72°C for 1 minute, followed by a final extension at 72°C for 10 minutes and a 4°C hold. All reactions were conducted in an Eppendorf mastercycler gradient machine. f Eight reactions were performed for each sample, and one product was resolved on a 1.5 agarose gel and stained with ethidium bromide to verify the presence of the 583-bp band. The remaining 7 amplified products from each sample were combined, and the amplicon was purified using an Ultrafree-DNA gel purification kit, g according to the manufacturer's instructions. Sequencing of purified amplicons was carried out by a commercial facility. h
Primers and probes used in this study.

For virus isolation, a 10% suspension of feces was prepared in DMEM supplemented with 10mM Hepes buffer and 1 μg/ml gentamicin sulfate. This suspension was clarified by centrifugation at 800 − g for 10 minutes, filtered through a 0.45-μ filter, and used to inoculate 2 cell lines: the A-72 canine kidney cells and Crandell-Reese Feline Kidney (CRFK). The inoculations were performed by adding 2 drops of inoculum onto freshly trypsinized cells seeded into 24-well plates at 1 ml/well (125,000 cells) in DMEM containing 10% fetal bovine serum. After 5 days of incubation at 37°C in 5% CO2, the cells were trypsinized, spotted into wells of Teflon-coated glass slides, fixed for 10 minutes in 100% acetone at room temperature, and stained for CPV antigen by reacting with a fluorescein isothiocyanate-labeled polyclonal anti-CPV antibody. i The cells from the first passage were reseeded into 24-well plates (2 wells per inoculum) and stained after 5 days if the first passage was negative for CPV. Cultures that remained negative after the second passage were scored as negative for virus isolation.

Nucleotide alignment of a VP2 fragment of American CPV-2c strains with reference strains CPV-2 CPVb (GenBank accession number M38245), CPV-2a CPV-15 (M24003), CPV-2b CPV-39 (M74849), CPV-2c 56/00 (AY380577), U51 (AY742942), and 695 (AF401519). Only the segment exhibiting differences is shown.
Of the 33 samples, 27 were found to be positive for CPV using real-time PCR. Table 1 lists the states of origin of the 33 samples and the MGB-PCR results for the 27 positive samples. Seven dog samples originating from 5 states were characterized as CPV-2c. The dogs were of 7 different breeds, ranged in age from 3 to 8 months, and had all received 2 or 3 doses of different types of commercially available CPV vaccines before onset of clinical signs, which included nausea, vomiting, and diarrhea. The single CPV-2a subtype and 17 of the 19 CPV-2b subtypes originated from clinically sick dogs of various breeds, ranging in age from 2 to 14 months, that had received 2 to 4 doses of various commercially available CPV vaccines. Using MGB-PCR analysis, 9 16 of the 27 CPV strains detected in this study (including the lone CPV-2a) were determined to be field strains rather than vaccine strains. The other 11 strains were not differentiated for vaccine or field origin. However, because all of these 11 strains were either CPV-2b or 2c, they could not be of vaccine origin because vaccines containing these subtypes are not used in the United States.
The PCR products of the 7 CPV-2c subtypes detected in this study were sequenced in the VP2 region spanning nucleotides 4002 through 4585 of the CPV genome and compared with GenBank sequences of CPV-2, CPV-2a, CPV-2b, and CPV-2c using a ClustalW-based online sequence alignment tool (http://justbio.com/aligner/index.php) (Fig. 1). Based on the occurrence of the codon GAA at position 4062 to 4064 (encoding for glutamine at residue 426), all 7 isolates were confirmed by sequencing as CPV-2c. However, the sequence alignment also showed that 4 of the 7 U.S. CPV-2c strains differed from 3 previously described European isolates by exhibiting 2 further mutations in the target region: GA at nucleotide 4076, which involves the third base of codon 430 and thus does not cause a change in residue; AG at nucleotide 4104, which affects the first nucleotide of codon 440, changing it from ACA to GCA, thus altering amino acid residue 440 from threonine to alanine (ThrAla). In the 2 states (Arizona and California) from which 2 CPV-2c were detected, one isolate in each case was identical to European isolates in the sequenced region, while the other exhibited the ThrAla mutation described in this study.
Virus isolation attempts yielded only 6 CPV isolates (3 CPV-2b and 3 CPV-2c). Identical results were obtained on both cell lines used (A-72 and CRFK). The relatively low sensitivity of virus isolation compared with real-time PCR has been previously reported 6,10 and could have been exacerbated in this study by the fact that several of the samples had been frozen and thawed multiple times, possibly diminishing or abrogating virus viability. Nevertheless, the availability of some isolates from this study provides material for further analysis and possible future use as vaccine or challenge-strain candidates.
The findings from this study indicate that CPV-2c is probably widely distributed in the United States, because the 7 isolates originated from geographically widely dispersed locations, and there was no evidence of prior contact between any of the dogs. It is not possible, however, to determine from this study how long CPV-2c might have occurred in the United States. The finding of mutations in 4 of the 7 U.S. isolates that do not occur in the 3 European CPV-2c isolates from 1997–2000, might mean that the 2 groups of CPV-2c isolates evolved independently. However, antigenic analysis and further sequence comparison involving more of the genome of both groups of isolates is required before a definitive statement can be made on their evolution.
Although it is tempting to think that CPV-2c could have emerged as an escape mutant from vaccination pressure, the results of this study do not seem to support that hypothesis. Indeed, most of the isolates typed in this study (19 of 27) were CPV-2b, and they originated from dogs with a vaccination history similar to the dogs from which CPV-2c was detected. Moreover, all 27 isolates originated from different dogs, and no dogs were infected with more than one subtype. Furthermore, although complete clinical histories were not obtained for all dogs, the short time interval (2 days) from vaccination to onset of clinical signs in 2 cases indicates that some of the dogs might have been infected before vaccine administration. Moreover, 2 of the dogs infected with CPV-2b had never received a CPV vaccine. It is also noteworthy that the 7 dogs infected with CPV-2c exhibited clinical signs and outcomes that were similar to those exhibited by the dogs infected with CPV-2a and CPV-2b.
The continuous evolution of CPV strains, coupled with persistent anecdotal reports of “vaccine breaks” may have important implications for vaccine formulations. Most vaccines used worldwide are based on the original 1978 CPV-2 dog isolate cultured at the James Baker Institute for Animal Research, New York State College of Veterinary Medicine (Cornell University, Ithaca, NY). Although there is evidence to show that vaccines based on this original CPV-2 isolate are protective against CPV-2c challenge (Toulemonde CE, Brunei S, Cariou C, et al.: 2006, Management of canine parvovirus type 2 in a kennel environment. Proc Journées GTV, Dijon, France; Spibey N, Greenwood N, Tarpey I, et al.: 2006, Canine parvovirus type 2 vaccine protects dogs following challenge with a recent type 2c strain. Proc World Small Animal Veterinary Association Congress, Prague), it is nonetheless important for pharmaceutical companies consider using current strains in vaccine formulations. 6,11,13,20,23 Such considerations recently led to the licensing of a CPV-2b-based vaccine in Europe. 13
The continuous evolution of CPV also has serious implications for diagnostic tests based on monoclonal antibodies and PCR. It has been shown that the Glu-426 change in CPV-2c results in an antigenic difference detectable by monoclonal antibodies 16 The ThrAla mutation detected in this study is located in close proximity to the Glu-426 residue in the major antigenic site (epitope A) found on the 3-fold spike of the CPV capsid protein 21 ; thus, it is possible that the ThrAla mutation could be antigenically significant, although this remains to be determined. Nevertheless, continuous evolution of CPV requires that diagnostic assays based on a single detecting monoclonal antibody be evaluated periodically for their continuous sensitivity against new strains of CPV. Similarly, nucleic acid-based tests need to be evaluated continuously to ensure that mutations have not occurred in primer/probe binding regions.
Acknowledgements. The technical help of Nate Chenoweth is greatly appreciated. This study was funded by a grant from Merial, Inc.
Footnotes
a.
UltraClean Fecal DNA Kit, MoBio Laboratories Inc., Carlsbad, CA.
b.
Bio-Rad Laboratories Sri, Milan, Italy.
b.
Bio-Rad Laboratories Sri, Milan, Italy.
c.
Bio-Rad Laboratories, Hercules, CA.
d.
Amplitaq GOLD Master Mix, Applied Biosystems, Branchburg, NJ.
e.
Primers and probes, IDT, Coralville, IA.
f.
Mastercycler Gradient, Eppendorf, Hamburg, Germany.
g.
Ultrafree-DA, Millipore, Bedford, MA.
h.
Oklahoma Medical Research Foundation, Oklahoma City, OK.
i.
Canine parvovirus direct fluorescent antibody conjugate, American Bioresearch, Sevierville, TN.