
Abstract
Sheep-associated malignant catarrhal fever (SA-MCF), a frequently fatal disease primarily of certain ruminants, is caused by ovine herpesvirus 2 (OvHV-2). Molecular diagnosis of SA-MCF in affected animals has relied on detection of OvHV-2 DNA using a nested PCR, which has significant potential for amplicon contamination as a routine method in diagnostic laboratories. In this report, a nonnested and a previously developed real-time PCR were validated for detection of OvHV-2 DNA in samples from clinically affected animals. Three sets of blood or tissue samples were collected: 1) 97 samples from 97 naturally affected animals with evidence of clinical SA-MCF; 2) 200 samples from 8 animals with experimentally induced SA-MCF; and 3) 100 samples from 100 animals without any evidence of clinical SA-MCF. Among 97 positive samples defined by nested PCR from clinically affected animals, 95 (98%) were positive by nonnested PCR and 93 (96%) were positive by real-time PCR, respectively. One hundred percent of the samples from the animals with experimentally induced MCF were positive by real-time PCR, while 99% were positive by nonnested PCR. Neither nonnested PCR nor real-time PCR yielded a positive result on any of the 100 nested PCR-negative samples from animals without evidence of clinical MCF. The data confirmed that both nonnested and real-time PCR maintained high specificity and sensitivity for the detection of OvHV-2 DNA in clinical samples.
Sheep-associated malignant catarrhal fever (SA-MCF), a herpesviral disease caused by ovine herpesvirus 2 (OvHV-2), can pose a significant diagnostic challenge to veterinary clinicians because the clinical presentation is difficult to differentiate from several other common viral diseases. 3 The classical signs of corneal edema, fever, and generalized lymphadenopathy are not always evident, particularly during the acute phase of disease in highly susceptible species such as deer and bison. 12 Although the predominant microscopic lesions of MCF (lymphoproliferation, mucosal inflammation, and vasculitis) are highly suggestive, they may either be absent or so mild 12 that the diagnostician may have difficulty in differentiating them from similar lesions that can accompany bovine viral diarrhea/mucosal disease, infectious bovine rhinotracheitis, and epizootic hemorrhagic disease. 3 Immunohistochemical or direct fluorescent antibody staining techniques have been unsuccessful, despite many attempts, in demonstrating of viral antigen in tissues. 4,14
PCR is one of the best nucleic acid-based molecular techniques for detection of specific pathogens or groups of pathogens in diagnostic laboratories. 17 The development of diagnostic reagents and techniques, including nucleic acid- based methods, for SA-MCF has been difficult because of a lack of sequence data. The cloning and sequencing of a fragment of the OvHV-2 genome 2 resulted in the development of a nested PCR assay specific for OvHV-2, 1 which advanced the ability to detect OvHV-2 DNA in animals with clinical MCF. 11 However, although highly sensitive, the use of nested PCR as a routine method to detect OvHV-2 DNA for confirmation of clinical SA-MCF in diagnostic laboratories is problematic because the nested amplification format has the potential for significant amplicon contamination. A real-time PCR for OvHV-2 recently developed by Hussy and coworkers 6 may become the test of choice, but performance of this assay for detection of OvHV-2 DNA in animals with clinical MCF has not been thoroughly evaluated. Using real-time PCR, preliminary data from the authors' laboratory revealed that there were high levels of OvHV-2 DNA ranging from hundreds to millions of copies per microgram of sample DNA in the blood and tissues of various OvHV-2 infected animals, including cattle, bison, deer, pigs, and other exotic species that naturally developed clinical MCF (Li, et al., unpublished data). 15 Additionally, the data from experiments of aerosol induction of MCF in bison and cattle showed that all animals had increased OvHV-2 DNA levels in peripheral blood before the onset of clinical signs, and virtually all tissues from experimentally infected bison and cattle that developed MCF contained a high level of OvHV-2 DNA (O'Toole, et al., unpublished data). 15 Based on these data, it was hypothesized that OvHV-2 DNA in the blood and tissues of ruminants with clinical MCF can be detected by the less-sensitive nonnested PCR and realtime PCR formats.
To validate nonnested PCR and real-time PCR for diagnosis of clinical SA-MCF in clinically susceptible species, 3 sets of tissue or blood samples were collected. The first set, consisting of 97 field clinical MCF samples from 97 animals, including bison (n = 67), cattle (n = 11), deer (n = 13), and a few other species (n = 6) (Table 1), was obtained from Washington Animal Disease Diagnostic Laboratory, Wyoming State Veterinary Laboratory, and other state veterinary laboratories. A field clinical MCF sample was defined by the following criteria: 1) the animal was suspected of having naturally acquired clinical MCF based on clinical signs and/or histological lesions that are typical of MCF, and 2) OvHV-2 DNA was detected in the blood or tissue sample from the animal by nested PCR. 8 The second set, consisting of 200 samples, was collected from 8 animals (7 bison and 1 calf) with experimentally induced MCF (O'Toole, et al., unpublished data). 15 These animals were experimentally aerosolized with various doses of infectious OvHV-2 collected from sheep nasal secretions. 16 All the nebulized animals developed clinical MCF, which was confirmed by histopathology and PCR specific for OvHV-2. Among the 200 samples, multiple tissue samples were collected from different locations in certain organs, such as turbinate, trachea, and lung from the respiratory tract of each individual animal. The lymph node samples included prescapular, retropharyngeal, and mesenteric lymph nodes. The third set of samples consisted of 100 nonclinical MCF samples from 100 animals, including 59 bison, 8 cattle, 13 deer, and 20 other species (Table 1). A nonclinical MCF sample was defined as being from an animal that was not suspected to have clinical MCF based on the absence of clinical signs and/or histological lesions and that was negative for OvHV-2 DNA by nested PCR. Total DNA was extracted from peripheral blood leukocytes (PBL) or tissues using the FastDNA kit a as described by the manufacturer. The protocol for the collection of PBLs from EDTA-blood was described previously. 10 The concentration of purified DNA was measured using a spectrophotometer at a wavelength of 260 nm.
The procedure for nonnested PCR was similar to the previously described nested protocol, 1,8 but used only the external set of primers: No. 556 (5′-AGTCTGGGGTATATGAATCCAGATGGCTCTC-3′)and No. 755 (5′-AAGATAAGCACCAGTTATGCATCTGATAAA-3′), which amplifies a 422 base pair (bp) DNA fragment. All amplifications were performed in 100 μl volume reactions containing 10 mM Tris-HCl (pH 8.0), 2 mM MgCl2, 200 μM dATP, dCTP, dGTP, dTTP, b 20 pM of each primer and 2 units Taq DNA polymerase. b Thermal cycling conditions were 5 minutes at 95°C, followed by 34 cycles of 94°C (1 minute), 60°C (1 minute), and 72°C (2 minutes), followed by a final extension for 7 minutes at 72°C. Each nonnested PCR test included a known positive control and a negative control (no target DNA). For product visualization, 10 μl of the amplified PCR products were analyzed by 2% agarose gel electrophoresis and ethidium bromide staining. Two different target DNA concentrations (500 ng and 50 ng) from each sample were tested, based on a pilot experiment using various target DNA concentrations including 2,000, 1,000, 500, and 50 ng, respectively, in each reaction from 10 randomly selected samples. In the pilot experiment, nonnested PCR amplified specific signals from all 10 samples and 9 samples when using 500 ng and 50 ng of target DNA, respectively, but specific amplifications were obtained from fewer samples when 1,000 ng and 2,000 ng of target DNA were used (data not shown).
Sample source for the SA-MCF nonnested and realtime PCR validation study.
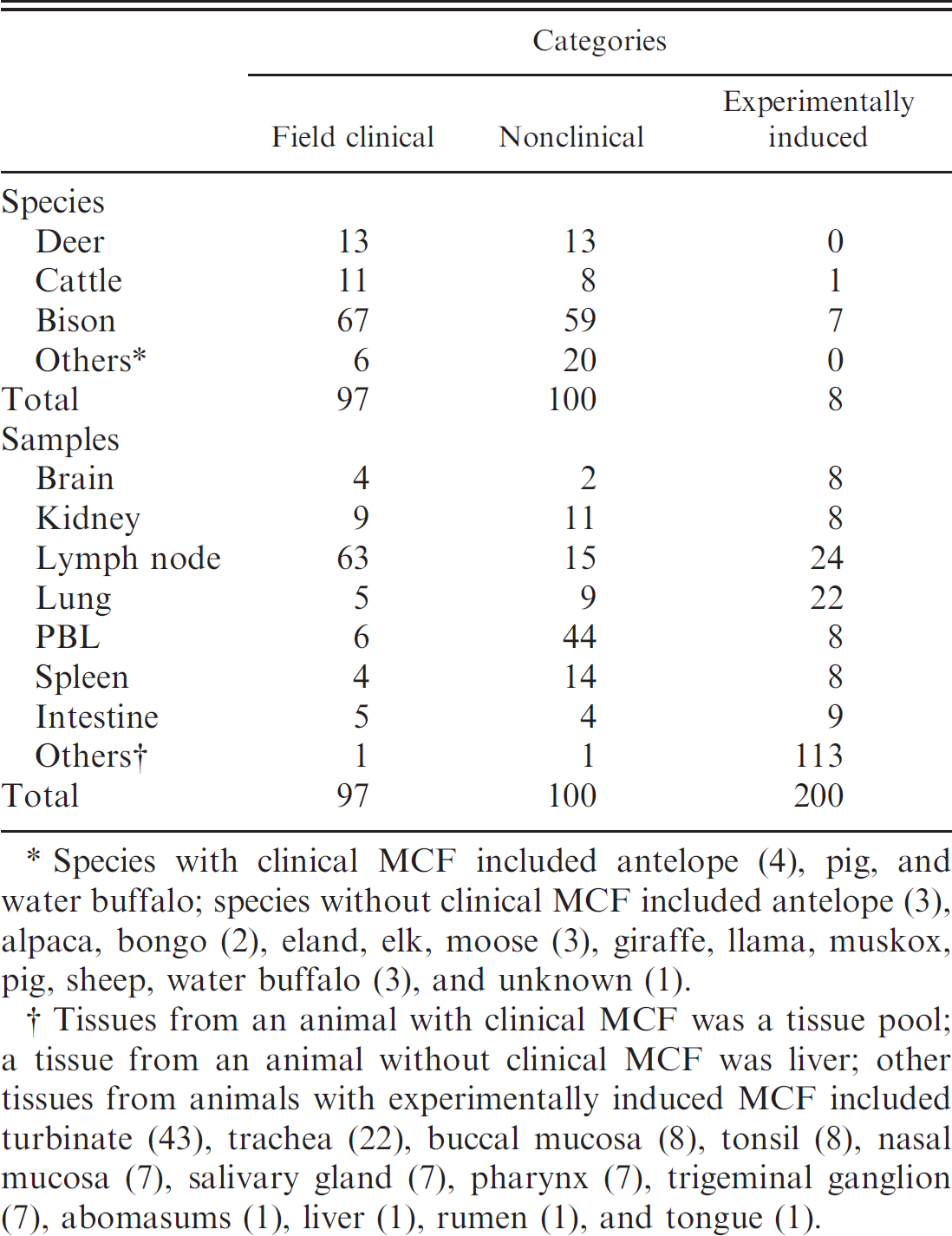
Species with clinical MCF included antelope (4), pig, and water buffalo; species without clinical MCF included antelope (3), alpaca, bongo (2), eland, elk, moose (3), giraffe, llama, muskox, pig, sheep, water buffalo (3), and unknown (1).
Tissues from an animal with clinical MCF was a tissue pool; a tissue from an animal without clinical MCF was liver; other tissues from animals with experimentally induced MCF included turbinate (43), trachea (22), buccal mucosa (8), tonsil (8), nasal mucosa (7), salivary gland (7), pharynx (7), trigeminal ganglion (7), abomasums (1), liver (1), rumen (1), and tongue (1).
The procedure for OvHV-2 real-time PCR was as previously described, 6,10 with forward primer (oF-OvHV-2, 5′-TGGTAGGAGCAGGCTACCGT-3′), reverse primer (oR-OvHV-2, 5′-ATCATGCTGACCCCTTGCAG-3′), and probe (oP-OvHV-2, 5′-TCCACGCCGTCCGCACTGTAAGA-3′), which was dual labeled with Fam at the 5′ end and Tamra at the 3′ end. Briefly, the 25 μl PCR mixture for one reaction contained 23 μl of TaqMan Universal Master Mix, c 240 nM forward primer (oF-OvHV-2), d 600 nM reverse primer (oR-OvHV-2), d 80 nM probe (oP-OvHV-2), c and 2 μl of diluted standard or template DNA. Both 500 ng and 50 ng of target DNA were tested for amplification, and each sample was amplified in duplicate. Seven serially diluted standards ranging from 101 to 107 OvHV-2 DNA copies and a no target DNA control were included in each test. Both the standard DNA and no DNA control were run in triplicate. The thermal cycling profile used was as follows: 95°C for 10 minutes to denature DNA and activate the AmpliTaq Gold DNA polymerase, followed by 40 cycles at 60°C for 1 minute and at 95°C for 15 seconds. Amplifications were performed in an iCycler iQ Real-Time PCR Detection System, e and data were analyzed using software (Version 2.3) e provided by the manufacturer.
Summary of the results from clinical samples tested by nonnested PCR and real-time PCR specific for OvHV-2.
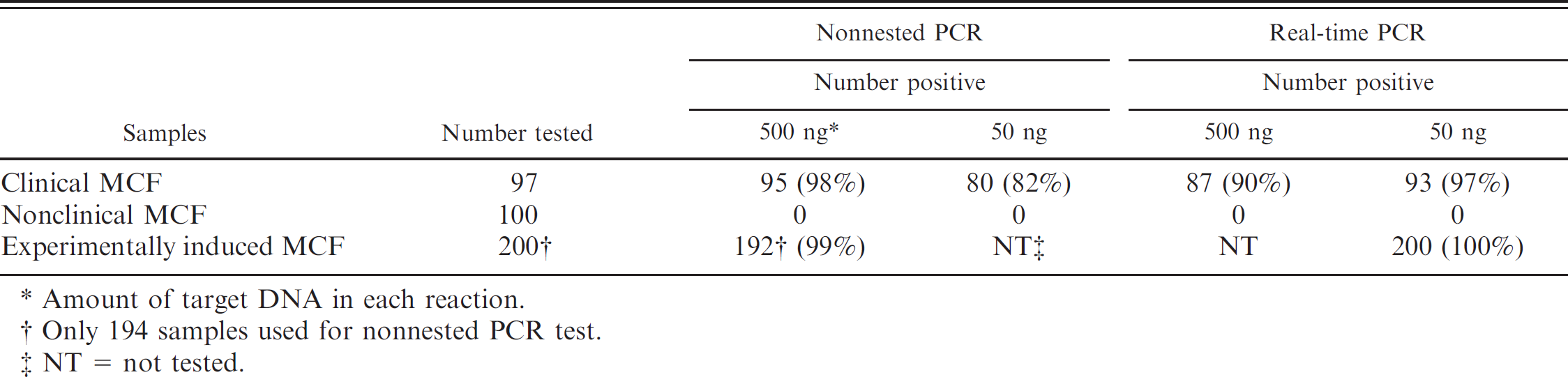
Amount of target DNA in each reaction.
Only 194 samples used for nonnested PCR test.
NT = not tested.
Among 97 field clinical MCF samples, 95 (98%) were positive by nonnested PCR using 500 ng of total DNA as an amplification target, whereas only 80 samples (82%) were positive when 50 ng of the target DNA was used (Table 2). Increasing target DNA to 1,000 ng or 2,000 ng per reaction did not increase detection sensitivity, but rather reduced the efficiency (data not shown), which most likely is due to increased PCR inhibitors in the reaction. This was also observed sometimes for the nested PCR assay when high concentrations of target DNA were used (Li, et al., unpublished data). When testing both concentrations of target DNA from these 97 field clinical MCF samples by real-time PCR, only 87 (90%) out of 97 had detectable copy numbers using 500 ng of target DNA, but 93 (97%) out of 97 were scored positive when 50 ng of target DNA was used (Table 2). The data suggest that the optimum concentration of target DNA differs in different PCR formats: 500 ng is optimal for the nonnested PCR assay, while 50 ng is optimal for the real-time PCR assay. The main reason for the effect of the target DNA concentration is likely due to the volume in the amplification reaction: a total 100 μl in nonnested PCR versus a total 25 μl in real time PCR. The larger volume likely dilutes out any PCR inhibitors that may be present.
The detailed comparison of real-time PCR results is shown in Table 3, including copy numbers and cycle threshold (CT) values, from both field (n = 93) and experimentally induced (n = 200) clinical MCF samples that were positive by real-time PCR using 50 ng of target DNA. The overall values, including mean, low, and high DNA copy numbers, were highly comparable between natural and experimental infections, which further suggest that these field clinical MCF samples were appropriately selected for the validation study. In addition, the data suggest that a CT value of 40 can be used for the cutoff in this real-time PCR based on the limited number of samples and the amplification conditions, because no false positives were observed within 40 cycles in the study.
Overall, the nonnested PCR and the real-time PCR were slightly less sensitive than the nested PCR. Current and previous data showed that the analytical sensitivity of the nonnested PCR assay using outer primers (556/755) at 34 cycles was approximately 300 OvHV-2 DNA copies, 5 and the real-time PCR assay was capable of detecting 50 copies, while the nested PCR assay was capable of detecting approximately 4 to 10 copies (data not shown).
The reasons for failure to detect OvHV-2 DNA in 4 field clinical MCF samples by either nonnested PCR (n = 2) or real-time PCR (n = 3) might be due to the levels of viral DNA in the samples and/or PCR inhibitors. One sample was negative by both nonnested PCR and real-time PCR, suggesting there were inadequate levels of OvHV-2 DNA in the sample, or it may have been a false-positive sample initially identified due to contamination in the nested PCR. The second sample that was negative by the nonnested PCR was positive by real-time PCR with a high level of viral DNA in the sample (77,600 copies/50 ng DNA). When tested using 50 ng of target DNA, it scored positive by nonnested PCR, suggesting that the viral DNA level was high in the sample, but PCR inhibitors became a problem when using an increased volume of sample to reach 500 ng of target DNA. No clear explanation could be given for the other 2 samples that were nonnested PCR-positive, but real-time PCR-negative; it might be owing to both relatively low viral copy numbers and higher concentration of PCR inhibitors in the samples. Neither nonnested PCR nor real-time PCR were positive for any of the 100 nonclinical MCF samples from animals that were not suspected of clinical MCF based on clinical signs or histological lesions and OvHV-2 negative by nested PCR (Table 2).
Comparison of real-time PCR results between the field clinical MCF samples and experimentally induced MCF samples.
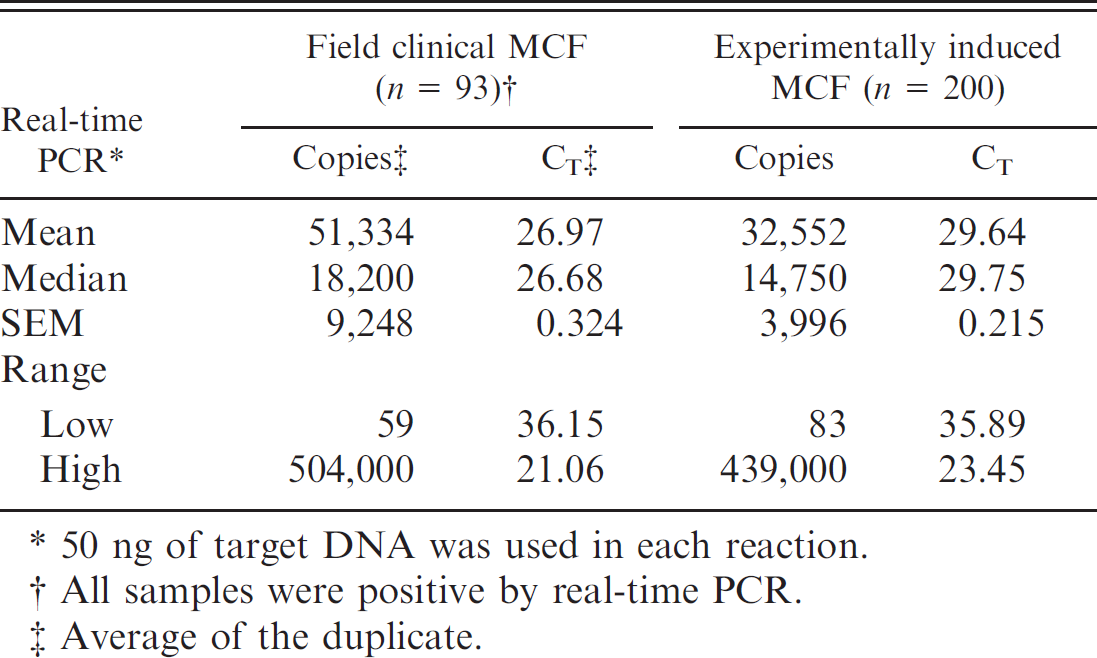
50 ng of target DNA was used in each reaction.
All samples were positive by real-time PCR.
Average of the duplicate.
The primary goal of this study was to determine whether nonnested PCR and real-time PCR were sensitive enough assays to detect OvHV-2 DNA in diagnostic samples from animals with SA-MCF. Among the 93 samples that were positive by real-time PCR, only 4 of them had viral DNA less than 100 copies in 50 ng of target DNA. It should be noted that clinically susceptible species, such as cattle or bison, can occasionally test positive by nested PCR without having clinical MCF. 12,13 In the 200 samples from animals with experimentally induced clinical MCF in this study, all except one had more than 100 OvHV-2 DNA copies in 50 ng of target DNA. This verified that OvHV-2 DNA in the blood and tissues of ruminants with clinical MCF could be detected by nonnested PCR and real-time PCR.
The nonnested PCR and real-time PCR validated herein can be used as reliable tools for detecting OvHV-2 DNA in samples from clinically affected ruminants. However, these assays may not be sensitive enough to detect OvHV-2 DNA in subclinically infected animals. Therefore, nested PCR must be used, especially when screening for early infection in lambs in order to produce OvHV-2-free sheep. 9 When OvHV-2 DNA levels are low, as in most subclinically infected bison and cattle, application of nested PCR for detection of infection may not be helpful. In such cases, serological assays, such as the competitive inhibition enzyme-linked immunosorbent assay (ELISA), 7 are useful. The presence of antibody is proof of infection, but not necessarily of clinical MCF, as a significant percentage of bison and cattle are subclinically infected. 12,13,15
AcknowledgementsThis work was supported in part by USDA/ARS CWU 5348-32000-024-00D. The authors thank Janice Keller, Lori Fuller, Shirley Elias, and Dan Bradway for technical assistance.
Footnotes
a.
QBiogene, Inc., Irvine, CA.
b.
Qiagen, Valencia, CA.
c.
Biosystems, Foster City, CA.
d.
d. Integrated DNA Technologies, Coralville, IA.
e.
e. Bio-Rad Laboratories, Hercules, CA.