
Abstract
Acute myeloid leukemia is the most common form of acute leukemia in adults, constituting about 80% of cases. Although remarkable progress has been made in the therapeutic scenario for patients with acute myeloid leukemia, research and development of new and effective anticancer agents to improve patient outcome and minimize toxicity is needed. In this study, the antitumor activity of axolotl (AXO) Ambystoma mexicanum crude extract was assessed in vitro on the human acute myeloid leukemia HL-60 cell line. The anticancer activity was evaluated in terms of ability to influence proliferative activity, cell viability, cell cycle arrest, and differentiation. Moreover, gene expression analysis was performed to evaluate the genes involved in the regulation of these processes. The AXO crude extract exhibited antiproliferative but not cytotoxic activities on HL-60 cells, with cell cycle arrest in the G0/G1 phase. Furthermore, the AXO-treated HL-60 cells showed an increase in both the percentage of nitroblue tetrazolium positive cells and the expression of CD11b, whereas the proportion of CD14-positive cells did not change, suggesting that extract is able to induce differentiation toward the granulocytic lineage. Finally, the treatment with AXO extract caused upregulation of CEBPA, CEBPB, CEBPE, SPI1, CDKN1A, and CDKN2C, and downregulation of c-MYC. Our data clearly show the potential anticancer activity of Ambystoma mexicanum on HL-60 cells and suggest that it could help develop promising therapeutic agents for the treatment of acute myeloid leukemia.
Introduction
Acute myeloid leukemia (AML) is a blood cancer characterized by the proliferation of clonal precursor myeloid cells with differentiation arrest. AML is the most common form of acute leukemia in adults constituting about 80% of cases. Its incidence is about 1.3 per 100,000 for those below 65 years and about 12.2 cases per 100,000 for those above 65. 1
At present, standard intervention for AML consists of chemotherapy and, secondarily, stem cell transplantation. However, in many cases, AML cells are not eradicated, with persistent cells reappearing after a period of remission. Also, the conventional therapies have often shown undesirable side effects (e.g. hepatotoxicity, myelosuppression, and tumor lysis syndrome). 2 Thus, it is urgent to find new therapeutic strategies for AML treatment.
Differentiation therapy, using agents that promote cancer cell differentiation, has been shown to be effective in in vitro and in vivo treatment of several types of cancers, including certain AML sub-types. 3
In recent years, significant emphasis has been placed on identifying new agents from natural sources that could be used against cancer. Natural products, extracted from abundant living organism sources, contain bioactive compounds that have the potential for prevention or treatment of major diseases. Many of these have been identified to demonstrate diverse biological actions, including anticancer activities. 4 Therefore, the screening of natural products against cancer is needed.
The axolotl Ambystoma mexicanum is one of the most widely used laboratory animals for stem cell research and regeneration. 5 The mechanisms governing regeneration, wound healing, development, and cancer formation are closely linked. There is increasing evidence highlighting the common signaling pathways which link cancer growth and regeneration whereby dysregulation of the well-balanced and coordinated process of regeneration leads to cancer. Thus, the identification of biologically active molecules from axolotl extracts could lead to major benefits, with directions on how to develop therapeutic approaches for cancer treatment in humans. Interestingly, Allegrucci et al. 6 found that axolotl oocyte extracts possess reprogramming ability, which reverses epigenetic silencing of tumor suppressor genes and tumorigenicity of breast cancer cells in a mouse xenograft model.
This study was planned to establish the potential anticancer activity of axolotl (AXO) Ambystoma mexicanum crude extract on human AML HL-60 cells. For this purpose, we evaluated the effect of AXO crude extract on cell proliferation, viability, cell cycle, and differentiation of HL-60 cells, and determined whether the AXO extract is able to modify the expression of genes involved in the regulation of these processes.
Materials and methods
Animals and tissue collection
The procedure was adapted from McGann et al. 7 Axolotls (A. mexicanum) were maintained in the laboratory at 21°C. Limb amputations were performed on animals anesthetized transdermally with 0.1% tricaine in water. A hindlimb was amputated by cutting just proximal to the elbow, and soft tissue was pushed up the humerus to expose the bone. The bone and soft tissue were trimmed to produce a flat amputation surface. After amputation, the axolotls were placed on ice for 1 h, transferred to a 0.5% sulfamerazine solution overnight, and then back into their normal water environment. After collection, all tissues were processed immediately for preparation of the wet extract.
Preparation of wet extracts
Tissues collected were kept on ice during the preparation of wet extracts. They were transferred to 50 mL tubes containing 10 mL sterile ice-cold Roswell Park Memorial Institute (RPMI; Biowest, Meda, Monza and Brianza, Italy) 1640 medium with 10% heat-inactivated fetal bovine serum (FBS; Biowest). Three protease inhibitors (2 µg/mL leupeptin/2 µg/mL aprotinin/1 mM phenylmethanesulfonyl fluoride (PMSF); Sigma-Aldrich, Milan, Italy) were added to the medium. The tissues were ground with an electronic tissue homogenizer for 1 to 2 min, hand homogenized in a sterile glass homogenizer for 10 to 15 min, and sonicated for 30 s in succession. The wet preparation homogenate was transferred to 2 mL microcentrifuge tubes, and the cell debris was removed in two centrifugation steps, the first being spun at 2000g for 25 min at 4°C. The supernatant was collected and centrifuged again at 100,000g for 60 min at 4°C. The final supernatant was filter-sterilized using a 0.22 µm filter and collected in a separate sterile 2 mL microcentrifuge tube. The protein content was assayed using the detergent compatible (DC) protein assay kit II (Bio-Rad Laboratories, Milan, Italy), and the tubes were stored in 1 mL aliquots in an −80°C freezer.
Cell culture
The human acute myeloid cell lines, HL-60 (ATCC-CCL-240) and KG-1a (ATCC-CCL-246.1), were supplied by the American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (Sigma-Aldrich). Cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was used to test the metabolic activity of the cells. HL-60 and KG-1a cells (1 × 105 cells/well) were seeded in 96-well plates and treated with different concentrations of AXO extract (0.25, 0.50, 1.00, and 2.00 mg/mL) for 24 to 72 h. MTT solution (5 mg/mL in phosphate-buffered saline (PBS); Sigma-Aldrich) was then added to each well and incubated for 4 h at 37°C, followed by the addition of 120 µL of dimethyl sulfoxide (DMSO). Finally, the plates were shaken and examined using a microplate reader by measuring at a certain wavelength (at 570 nm, with 690 nm as reference wavelength). All experiments were repeated at least three times, and each assay was performed in triplicate. Metabolic activity was calculated as follows: Metabolic activity (%) = (OD value of experimental samples/optical density [OD] value of control samples) × 100%.
Cell proliferation and viability by Trypan blue assay
HL-60 cells (1 × 105/well) were seeded in 48-well plates and treated with different concentrations (0.25, 0.50, 1.00, and 2.00 mg/mL) of AXO crude extract or 10% of DMSO (Sigma-Aldrich) for 24 to 72 h. Next, total cell number and viability were evaluated by Trypan blue exclusion assay using Countess II FL Automated Cell Counter (Invitrogen by Thermo Fisher Scientific, Waltham, MA, USA). Live cells excluded the dye, whereas dead cells admitted the dye intensely staining with Trypan blue.
Leishman staining
Cells (2 × 104/well) were seeded in 96-well plates and treated with AXO crude extract (2.0 mg/mL), DMSO (1.6%), or phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) (10 nM) for 72 h. Next, cells were collected on slides prepared by cytospin centrifugation at 200g for 5 min and stained using Leishman stain (Sigma-Aldrich) according to the manufacturer’s protocol. The morphological analysis was performed using the EVOS FL Auto 2 Cell Imaging System (Invitrogen).
Analysis of cell cycle by flow cytometry
HL-60 cells (1 × 105/well) were seeded in 48-well plates and treated with AXO crude extract (2.0 mg/mL), DMSO (1.6% and 10%), or PMA (10 nM) for 72 h. The cell cycle phase distribution was studied by quantitation of DNA content. Cells were collected, centrifuged, washed with 1× PBS, and fixed with 70% ethanol overnight at 4°C. Then, after removing ethanol and washing twice with PBS, cells were exposed to 100 µL RNase A (100 µg/mL; Sigma-Aldrich) for 1 h at 37°C, and stained with 200 µL propidium iodide (PI) (100 µg/mL; Sigma-Aldrich) in the dark for 15 min at 4°C. The DNA content and cell cycle distribution were analyzed by FACSCalibur flow cytometer (Becton Dickinson, Oxford, UK), and the data were analyzed utilizing Cell Quest software. At least 1 × 104 events were acquired for each sample.
Nitroblue tetrazolium reduction assay
HL-60 cells (2 × 104/well) were seeded in 96-well plates and treated with AXO crude extract (2.0 mg/mL) or DMSO (1.6%, as a positive control) for 72 h. Next, cells were incubated with 2 mg/mL nitroblue tetrazolium (NBT) solution (Sigma-Aldrich) at 37°C for 30 min. Then, the percentage of cells containing intracellular blue–black formazan deposits was determined. In each count, at least 200 cells were inspected using the EVOS FL Auto 2 Cell Imaging System.
Detection of CD11b and CD14 by flow cytometry
HL-60 cells (1 × 105 cells/well) were seeded in 48-well plates and exposed to AXO crude extract (2.0 mg/mL), DMSO (1.6%), or PMA (10 nM) for 72 h. Cells were collected, centrifuged, washed twice with precooled PBS, and incubated with direct Alexa Fluor 488-labeled anti-CD11b antibody or phycoerythrin (PE)-labeled anti-CD14 antibody (BD Pharmingen, Buccinasco, Milan, Italy) on ice on rotating platform in the dark. After 30 min, cells were washed twice and analyzed by FACSCalibur flow cytometer (Becton Dickinson), and the data were analyzed utilizing Cell Quest software. At least 1 × 104 events were acquired for each sample. Expression of cell marker was determined by comparison with isotype control signal.
RNA extraction and real-time reverse transcription polymerase chain reaction
Total RNA was isolated using RNeasy Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The amount of RNA extracted was determined using the Nanodrop 2000 instrument (Thermo Fisher Scientific). Complementary DNAs (cDNAs) were produced from 300 ng of RNA from each sample using the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using TaqMan™ Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) (Table 1). Two microliters of the diluted cDNA were amplified in a final volume of 25 µL with 1× TaqMan™ Gene Expression Master Mix (Applied Biosystems). Thermal cycling consisted of 2 min at 95°C, 40 cycles at 95°C for 15 s, and 60°C for 1 min. All real-time PCR reactions are performed in triplicate. No reverse transcriptase (no RT) control and a cDNA free negative control were included during each run. Real-time PCR and data collection were performed on Bio-Rad CFX96 System Cycler Instrument (Bio-Rad Laboratories). The relative expressions of mRNAs were calculated using the comparative 2–ΔΔCt method 8 and normalized against the geometric average expression of HPRT1 and YWHAZ housekeeping genes.
TaqMan® Gene Expression Assays.

Statistical analysis
All experiments were performed in triplicate and repeated at least twice. The SPSS software (version 20) was used for statistical analysis. Data are presented as the median and the interquartile range (IQR). Normal distribution was assessed by the Kolmogorov–Smirnov test. Since data were not normally distributed, they were analyzed using the nonparametric Kruskal–Wallis test, followed by Dunn–Bonferroni’s post hoc analysis. Significance was accepted at p < 0.05.
Results
Effects of the AXO extract on metabolic activity of HL-60 and KG-1a cells
First, the effects of AXO crude extract were tested on the metabolic activity of HL-60 and KG-1a cells using the MTT assay. Both cell lines were treated for various times (24–72 h) with various doses of AXO extract (0.25–2.00 mg/mL). DMSO (10%) was used as a positive cytotoxic control.
As shown in Figure 1(a), AXO extract significantly reduced HL-60 metabolic cell activity from 24 to 72 h compared to untreated cells. In fact, after 72 h treatment, metabolic activity was reduced between 40% and 50%. However, AXO extract slightly reduced KG-1a metabolic cell activity at 72 h at a concentration of 2.0 mg/mL (Figure 1(b)). In fact, after 72 h treatment, metabolic activity was reduced less than 20%. Since preliminary data showed that AXO extract is particularly effective on HL-60 cells, we decided to carry further investigations using the HL-60 cell line.

Effects of the axolotl (AXO) Ambystoma mexicanum crude extract on metabolic activity of (a) HL-60 and (b) KG-1a cells. Cells were treated with AXO or DMSO extract as indicated for 72 h. Metabolic activity was calculated and data are presented as median and interquartile range (error bars; n = 3 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05; **p < 0.001).
AXO extract reduces HL-60 cell proliferation
The antiproliferative effects of AXO extract on HL-60 cells were also evaluated by the Trypan blue assay. As shown in Figure 2(a) and (b), AXO extract significantly reduced the cell number of HL-60 cells after 48 h. At 72 h, a dose-dependent effect was observed with growth inhibition of 15%, 24%, 32%, and 50% for 0.25, 0.50, 1.00, and 2.00 mg/mL, respectively, compared to untreated cells. We also found that growth inhibition caused by AXO extract treatment is not due to decreased cell viability (Figure 2(c)).

Effects of the axolotl Ambystoma mexicanum (AXO) crude extract on (a) cell number, (b) relative cell growth (percentage of cell number of treated cells relative to untreated cells), and (c) cell viability of HL-60 cells. Cells were treated with AXO extract as indicated for 24, 48, and 72 h. The number of viable cells was determined by Trypan blue exclusion. Data are presented as median and interquartile range (error bars; n = 2 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05; **p < 0.001).
Based on the data obtained, to investigate the possible anticancer effects of the A. mexicanum extract, we decided to perform all the subsequent experiments by incubating HL-60 cells with the concentration of 2.0 mg/mL for 72 h.
HL-60 cells treated with AXO extract show G0/G1 arrest
HL-60 cells incubated with DMSO (1.6%) and PMA (10 nM) were used as positive controls for differentiation, as well as with AXO extract. As shown in Figure 3(a) and (b), flow cytometry analysis indicated that among the untreated cells, 58.2% were distributed in G0/G1 phase, 26.4% were accumulated in S phase, 13.1% were in G2/M phase, and only 2.3% were in sub-G0/G1 phase. Incubation of HL-60 cells with AXO extract (2.0 mg/mL) for 72 h resulted in a marked increase of cells in the G0/G1 phase (75.1%), with concomitant loss of S (16.8%) and G2/M (7%) phases.
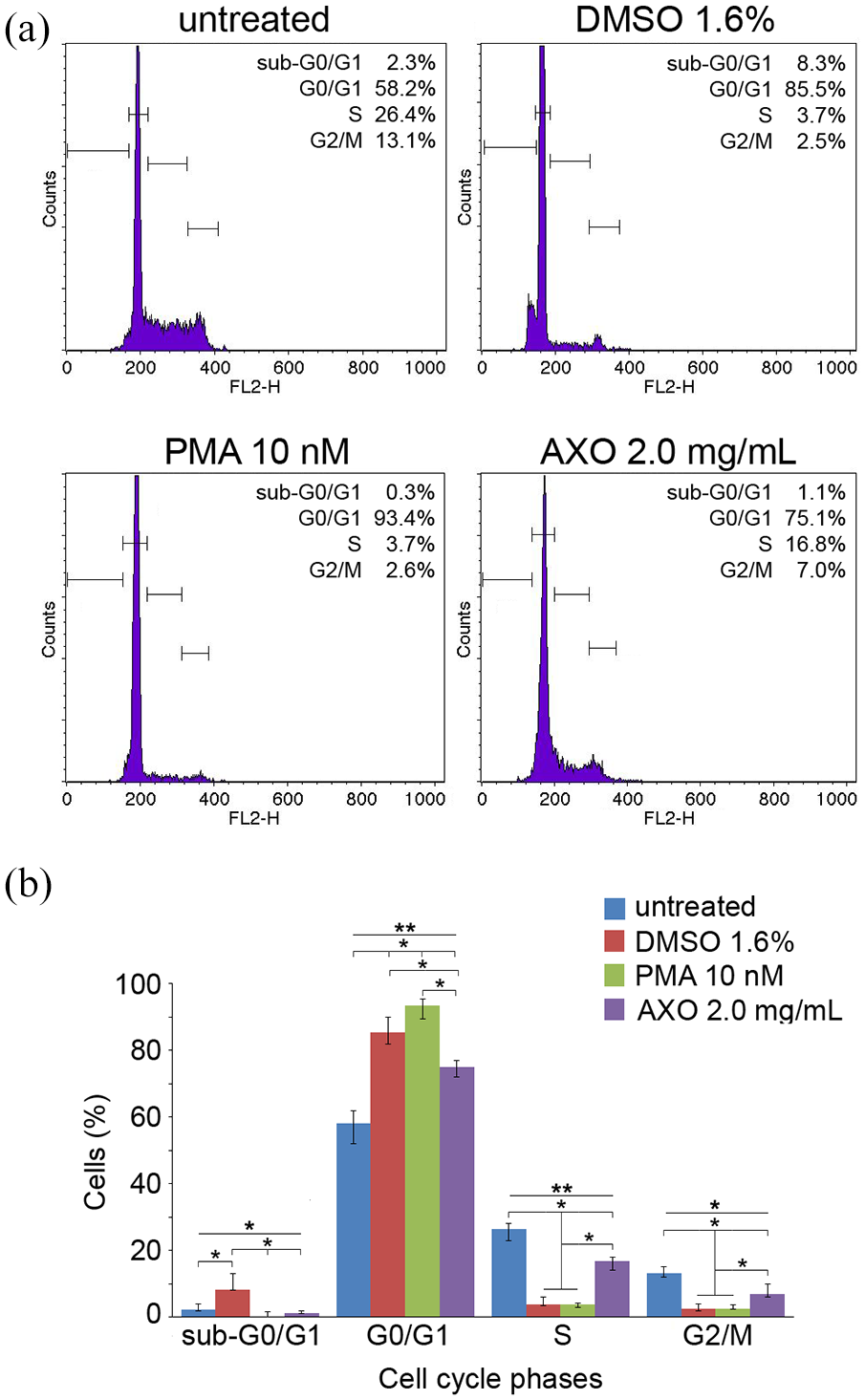
Effects of AXO extract (2.0 mg/mL) treatment on cell cycle distribution in HL-60 cells after 72 h. (a) Histogram plots of flow cytometry analysis performed in HL-60 cells. (b) Graph summarizing the cell cycle distribution. DMSO (1.6%) and PMA (10 nM) were used as positive control for granulocytic and monocytic differentiation, respectively. Data are presented as median and interquartile range (error bars; n = 2 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05; **p < 0.001).
AXO extract induces partial granulocytic differentiation in HL-60 cells
The morphologic changes of cells treated with AXO extract were viewed by Leishman staining. HL-60 cells incubated with DMSO (1.6%) and PMA (10 nM) were used as positive controls for granulocytic and monocytic differentiation, respectively. Untreated cells showed typical myeloid leukemia cell morphology with a small nucleus/cytoplasm (N/C) ratio and a small rim of basophilic cytoplasm, while AXO extract (2.0 mg/mL) increased the N/C ratio in HL-60 cells at 72 h (Figure 4(a)). The NBT assay was performed to detect the production of oxidative burst enzyme expression during the cell differentiation. As shown in Figure 4(b), NBT positive cells were increased after treatment by AXO extract (2.0 mg/mL) for 72 h.

AXO extract changes the morphology of HL-60 cells. (a) Leishman staining. The scale bar is 100 µm. The images were a representative of three independent experiments. DMSO (1.6%) and PMA (10 nM) were used as positive controls for granulocytic and monocytic differentiation, respectively. (b) Nitroblue tetrazolium (NBT) reduction assay. Cells were treated with AXO extract (2.0 mg/mL) for 72 h. HL-60 cells incubated with DMSO (1.6%) were used as positive controls for granulocytic differentiation. Graph summarizes the percentages of NBT positive cells. Data are presented as median and interquartile range (error bars; n = 2 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05).
To further characterize the differentiation mediated by AXO extract, flow cytometry analysis for myelomonocytic markers CD11b and CD14 was performed. HL-60 cells incubated with DMSO (1.6%) and PMA (10 nM) were used as positive controls for granulocytic and monocytic differentiation, respectively. An increase in the presence of CD11b-positive cells was observed in HL-60 cells treated with AXO extract (2.0 mg/mL) for 72 h, whereas the proportion of CD14-positive cells did not change (Figure 5(a) and (b)). These findings suggest that AXO extract is inducing granulocytic differentiation in HL-60 cells in vitro.

AXO extract–induced cell differentiation in HL-60 cells. (a) Cytometric analyses showing cell surface expression of CD11b (left panels) and CD14 (right panels) in HL-60 cells treated with DMSO (1.6%), PMA (10 nM), and AXO extract (2.0 mg/mL) for 72 h. (b) Graph summarizing both CD11b and CD14 reactivity. Data are presented as median and interquartile range (error bars; n = 2 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05; ns: not significant).
Effects of AXO extract treatment on gene expression in HL-60 cells
To investigate the molecular mechanisms by which AXO extract may alter the cell cycle progression and the differentiation state of HL-60 cells, the expression of genes involved in the regulation of differentiation (CEBPA, CEBPB, CEBPE, SPI1, and c-MYC) and cell cycle (CDKN1A and CDKN2C) was quantified by real-time RT-PCR analysis. HL-60 cells incubated with DMSO (1.6%) were used as positive controls for granulocytic differentiation.
As shown in Figure 6(a) and (b), compared to untreated counterpart, the treatment with AXO extract (2.0 mg/mL) increased the expressions of CEBPA, CEBPB, CEBPE, SPI1, CDKN1A, and CDKN2C, whereas it strongly reduced the expression of c-MYC after 48 h. Figure 6(a) and (b) also shows that, after 72 h, the treatment with AXO extract further increased the expressions of CEBPE and CDKN2C.
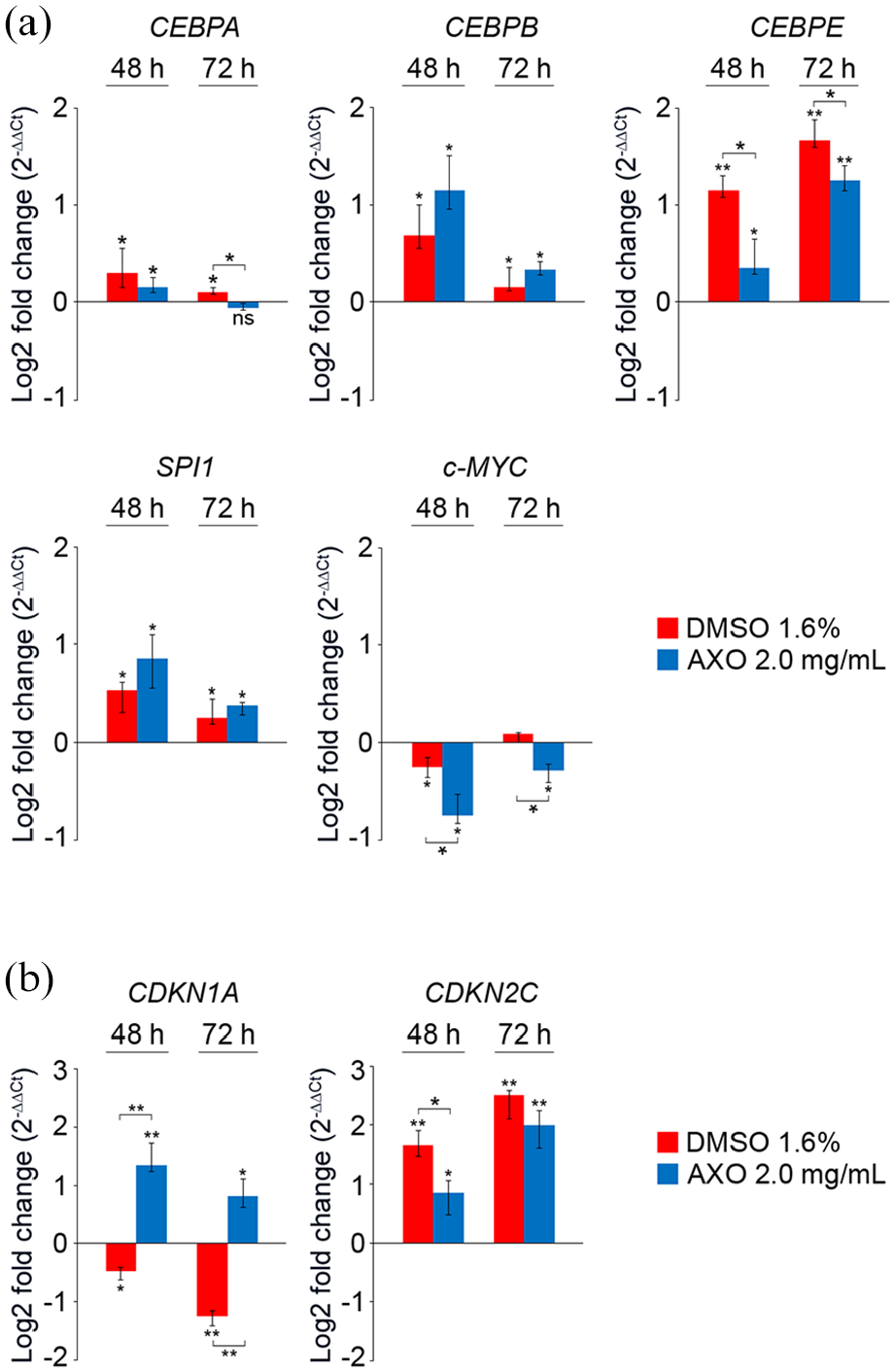
Differentially expression genes in response to AXO extract treatment in HL-60 cells. Cells were treated with AXO extract (2.0 mg/mL) for indicated times. mRNA expression of genes involved in (a) cell differentiation and (b) cell cycle regulation was determined by real-time RT-PCR. HL-60 cells incubated with DMSO (1.6%) were also used as positive controls for granulocytic differentiation. Relative transcript levels were determined using the 2–ΔΔCt method and normalized to HPRT1 and YWHAZ housekeeping genes. Expressions in untreated cells were used as calibrators. Data are presented as median and interquartile range (error bars; n = 2 independent experiments carried out in triplicate), and were analyzed using nonparametric Kruskal–Wallis, followed by Dunn–Bonferroni’s post hoc analysis (*p < 0.05; **p < 0.001).
Discussion
In this study, the AXO crude extract exhibited differentiation-inducing activity in human HL-60 cells, a model system for studying human myeloid cell differentiation. Our results suggest that AXO extract can inhibit cell growth of HL-60 cells probably via inducing G0/G1 phase arrest. Also, A. mexicanum extract induces partial granulocytic morphological changes in HL-60 cells.
Morphologically, HL-60 cells treated with AXO resembled the appearance of “paramyeloid” cells with a granulocyte-like nucleus but monocyte-like cytoplasm similar to those described by Zinzar et al. 9 These cells were also described in patients with chronic myelomonocytic leukemia. 10 The cells lacked nuclear indentations and resembled “partially differentiated” HL-60 cells following treatment with arsenic trioxide. 11 The morphological changes including chromatin condensation and decreased nucleocytoplasmic ratio after AXO treatment are similar to those seen following treatment with organic plant extracts such as coumarin, arsantin (a sesquiterpene lactone compound present in Artemisia santolina), and the differentiation-inducing agent, all-trans retinoic acid (ATRA).12–14
AXO-treated cells resemble early committed precursors and as such would continue to divide before acquiring a fully differentiated state. This is reflected in the reduced rate of cell proliferation when compared to untreated cells and an increase in the G0/G1 peak but without complete inhibition of proliferation. Brown et al. 15 similarly reported that HL-60 cells treated with ATRA committed to differentiation before cell cycle arrest, with differentiated cells not restricted to any particular stage of the cell cycle. These cells showed elements of innate immune responses, concomitant with the acquisition of differentiated function despite ongoing expansion of cell numbers. 16 Such gain in immune function of leukemia cells would be a benefit to neutropenic patients with the disease. Moreover, HL-60 cells induced to differentiate in response to ATRA expressed the early differentiation marker CD11b before halting cell division. 17
The molecular mechanisms by which AXO extract interferes with the cell cycle and induces differentiation of HL-60 cells are yet unknown; however, differentiation of HL-60 cells following treatment with AXO extract increased expression of CDKN1A and CDKN2C. Schwaller et al. 18 found that granulocytic differentiation induced by DMSO was accompanied by an increase in the CDKN2C protein product p18 expression, suggesting a direct role of this CDKi during myeloid differentiation. Horie et al. 19 showed that increased expression of p21, the protein product of CDKN1A, is necessary for the differentiation of HL-60 cells induced by ATRA. In our study, AXO strongly induces CDKN1A, like ATRA-induced differentiation but differently from DMSO-induced differentiation. We speculate that AXO, similar to ATRA and DMSO, induces granulocytic differentiation via the mitogen-activated protein kinase (MAPK) signaling pathways. Previous studies have revealed that both ATRA and DMSO induce HL-60 cells to differentiate into granulocytes via the extracellular-signal-regulated kinase (ERK) phosphorylation signaling pathway. These reports indicated that ERK1/2 phosphorylation is required to differentiate HL-60 cells into granulocytes and macrophages. 20
Importantly, AXO is inducing CDKN1A expression in a p53-independent manner, as HL-60 cells lack a functional p53 gene. 19 This implies that AXO may provide means for relative cell cycle arrest and activation of differentiation via CDKN1A in p53-null tumor cells. Interestingly, p53-null tumor cells usually respond less efficiently to direct cytotoxic chemotherapy. 21
Expression studies of myeloid transcription factors, here, complement the morphological and surface marker picture, with AXO-exposed cells exhibiting a granulocyte-like gene signature. Marchwicka and Marcinkowska 22 showed that a strong and sustained expression of CEBPB, when accompanied by the sustained expression of CEBPE, leads to granulocytic differentiation, 23 as also seen here. AXO-treated HL-60 cells resulted in overexpression of SPI1 (PU.1) and c-MYC repression. The downregulation of c-MYC by several compounds, including nicotinamide, coumarin, and 4-hydroxyquinazoline, causes arrest of leukemia cell proliferation and ultimately sustains myeloid differentiation of AML cells. 24 Taken together, our findings show that AXO crude extract exhibits antiproliferative and pro-differentiation action on HL-60 cells (Figure 7).

Schematic representation of the effects of Ambystoma mexicanum crude extract in HL-60 cells. Possible mechanisms include inducing cell differentiation and reducing cell proliferation. Green arrows represent increased expression, red arrows indicate decreased expression, and black double-headed arrows indicate unchanged expression.
Based on our results and previous studies, 25 we hypothesize that AXO extract might contain biologically active molecules similar to those derived from other amphibians including biogenic amines, bufodienolides (bufogenines and bufotoxins), alkaloids, peptides, and proteins that are potentially useful as anticancer agents. Interestingly, similar to AXO extract, the steroid bufalin (isolated from amphibian skin) seems to reduce the expression of c-MYC in HL-60 cells. Whereas reduction of c-MYC in AXO results in cell differentiation, bufalin induces apoptosis in HL-60 cells. Further characterization of the extract in our study might lead to identifying the active molecules, analyzing their specific pharmacological and toxicological effects, and the functional molecular mechanisms involved, especially in “in vivo” models. 26
Currently, isolating bioactive compounds from natural sources for producing pharmaceutical agents has become the focus of much attention. In particular, these compounds may act as promising sources for novel therapeutic agents in the treatment of human cancers. 4 Novel anticancer agents may arrest the cell cycle and induce apoptosis and/or stimulate differentiation.27–30 AXO extract holds a lot of potential, particularly in AML.
Footnotes
Acknowledgements
The authors wish to thank Professor Neville Calleja, from Public Health Department, University of Malta, Msida, Malta; Directorate for Health Information and Research, Gwardamanga, Malta, for professional assistance with the statistic analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by a research grant from the University of Malta. The sub-project numbers are ANARP16-17 and ANARP17-17.
Ethical approval
All procedures performed in this study involving animals were covered by ethical approval from the University of Malta Research Ethics Committee, Ref. No.: FRECMDS_1819_002.