
Abstract
Chronic myeloid leukemia is characterized by the presence of the reciprocal translocation t(9;22) and the BCR/ABL oncogene. The BCR/ABL oncogene activates multiple signaling pathways and involves the dysregulation of oncogenes during the progression of chronic myeloid leukemia. The cell division cycle protein 6, an essential regulator of DNA replication, is elevated in some human cancer cells. However, the expression of cell division cycle protein 6 in chronic myeloid leukemia and the underlying regulatory mechanism remain to be elucidated. In this study, our data showed that cell division cycle protein 6 expression was significantly upregulated in primary chronic myeloid leukemia cells and the chronic myeloid leukemia cell line K562 cells, as compared to the normal bone marrow mononuclear cells. BCR/ABL kinase inhibitor STI571 or BCR/ABL small interfering RNA could significantly downregulate cell division cycle protein 6 messenger RNA expression in K562 cells. Moreover, phosphoinositide 3-kinase/AKT pathway inhibitor LY294002 and Janus kinase/signal transducer and activator of transcription pathway inhibitor AG490 could downregulate cell division cycle protein 6 expression in K562 cells, but not RAS/mitogen-activated protein kinase pathway inhibitor PD98059 had such effect. Cell division cycle protein 6 gene silencing by small interfering RNA effectively resulted in decrease of proliferation, increase of apoptosis, and arrest of cell cycle in K562 cells. These findings have demonstrated that cell division cycle protein 6 overexpression may contribute to the high proliferation and low apoptosis in chronic myeloid leukemia cells and can be regulated by BCR/ABL signal transduction through downstream phosphoinositide 3-kinase/Akt and Janus kinase/signal transducer and activator of transcription pathways, suggesting cell division cycle protein 6 as a potential therapeutic target in chronic myeloid leukemia.
Introduction
The cell division cycle protein 6 (Cdc6), as a key regulator of DNA replication, plays a central role in the assembly of pre-replicative complexes (pre-RCs) at the origin of replication during the G1 phase of the cell division cycle.1,2 In early G1, Cdc6 is the first factor to be recruited by the origin recognition complex (ORC), and this binding of Cdc6 to ORC is a precondition for the loading of minichromosome maintenance (MCM) proteins onto chromatin.3,4 DNA is licensed for replication during the G phase once MCM proteins are recruited to pre-RCs. This involves the assembly onto chromatin of pre-RCs containing the replication licensing factors ORC, Cdc6, Cdt1, and MCM2–7, thereby rendering origins licensed for one round of DNA replication during S phase. 5 Several lines of evidence also indicate that Cdc18/Cdc6 are required to activate the checkpoint kinase Cds1 (Chk2/Rad53) and maintain the block to mitosis in S-phase-arrested cells.6,7 Loss of Cdc6 expression has been shown to cause a loss of chromosomal material and G2/M checkpoint control resulting in abnormal daughter cells.8,9 These studies suggest that Cdc6 plays important roles in the activation and maintenance of the checkpoint mechanisms that coordinate S-phase and mitosis.
Although Cdc6 is an important component of the cell-cycle regulatory mechanism, its potential involvement in carcinogenesis had not been investigated until recently. Studies show that Cdc6 is associated with oncogenic activity in some human cancers. High levels of Cdc6 protein have been reported in cervical cancer, brain tumors, non-small-cell lung carcinomas (NSCLCs), and epithelial ovarian cancer.10–13 Cdc6 overexpression interferes with the expression of INK4/ARF tumor suppressor genes through a mechanism involving the epigenetic modification of chromatin at the INK4/ARF locus.14,15 These studies suggest a possible role of Cdc6 in carcinogenesis and cancer progression. However, it is not clear yet whether Cdc6 expression is associated with the initiation, promotion, and progression in chronic myeloid leukemia (CML).
CML is a myeloproliferative disease characterized by the presence of the reciprocal translocation t(9;22) and the BCR/ABL oncogene.16,17 BCR/ABL activates multiple signaling pathways leading to protection from apoptosis, induction of growth factor–independent proliferation, modulation of adhesion/invasion ability, and induction of resistance to drugs. 18 BCR/ABL fusion protein, a constitutive tyrosine kinase, activates the phosphoinositide 3-kinase (PI3K)/Akt, Ras/Raf/mitogen-activated protein kinase (MAPK)/extracellular signal–regulated protein kinase (ERK), and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signal transduction pathways during the progression of CML.19,20 Despite substantial progress in the identification of the protein effectors of BCR/ABL kinase, little is known about the role of human Cdc6 gene in the BCR/ABL signal transduction pathways. So, examination of Cdc6 gene expression and signaling pathways may lead to a better understanding of the biology of BCR/ABL-positive leukemia and help identify potential targets for therapeutic applications.
In this study, we investigated the expression of Cdc6 in human CML cells, and the effect of the inhibition of Cdc6 expression by Cdc6 small interfering RNA (siRNA) on cell proliferation and apoptosis in K562 cells. Moreover, we evaluated the contribution of BCR/ABL and related signal transduction pathways to Cdc6 expression in CML cells. Our aim was to provide mechanistic evidence for the role of Cdc6 as BCR/ABL-dependent survival factor in the progression of CML. Our findings suggested that Cdc6 could be a potential target gene for the treatment of CML.
Materials and methods
Cell culture
The CML-derived cell line K562 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in complete medium (RPMI 1640 medium containing 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum (FBS)) at 37°C in a 5% CO2 incubator and resuspended in fresh culture medium every 2 days. The experiments were performed on cells during their exponential phase of growth. Bone marrow samples were obtained from patients presenting with CML and healthy volunteers at Union Hospital. Mononuclear cells were isolated from bone marrow samples by density centrifugation using Ficoll and cultured under the same conditions as used for growing K562 cells. Almost all CML bone marrow mononuclear cells or K562 cells had a single Ph chromosome as determined by fluorescence in situ hybridization (FISH) analysis. FISH was performed according to the manufacturer’s instructions using the Vysis LSI BCR/ABL Dual Color, Dual Fusion Translocation Probe (Abbott Molecular, Abbott Park, IL, USA). The BCR/ABL probe is a mixture of the LSI BCR probe labeled with spectrum green and the LSI ABL probe labeled with spectrum orange. Ethics approval for this study was provided by Ethics Committee of Union Hospital of Tongji Medical College, Huazhong University of Science and Technology. All participants provided written informed consent.
Treatment with inhibitors
Cell lines or primary cells were incubated with the PI3-kinase inhibitor LY294002 (20 µmol/L), the MAPK/ERK (MEK) inhibitor PD98059 (50 µmol/L), the JAK/STAT inhibitor AG490 (50 µmol/L; all from Calbiochem, San Diego, CA, USA), the BCR/ABL kinase inhibitor STI571 (0.5, 1.0, and 2.0 µmol/L; provided by Novartis Pharma AG, Basel, Switzerland), or control medium with 10% FBS at 37°C for up to 12 h.
Messenger RNA extraction and quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (RT-PCR) assays were used to determine the expression of Cdc6 messenger RNA (mRNA) and performed using the SYBR Green PCR Master mix on an automatic MX3000P real-time RT-PCR thermal cycler (Stratagene, La Jolla, CA, USA). Total RNA of the K562, CML, and normal bone marrow mononuclear cells was extracted using the RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. Total RNA (1 µg) was reversely transcribed to complementary DNA (cDNA) using a Moloney murine leukemia virus (M-MLV) Reverse Transcriptase kit (Invitrogen, Carlsbad, CA, USA). The primers for Cdc6 were as follows: 5′-gagccaagaaggagcacaagatt-3′; 5′-cttccaagagccctgaaagtgac-3′. The primers for BCR/ABL were as follows: 5′-ggagctgcagatgctgaccaac-3′; 5′-tcagaccctgaggctcaaagt-3′. The primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA (designed with Primer 5.0) were as follows: 5′-gaaggtgaaggtcggagtc-3′; 5′-gaagatggtgatgggatttc-3′. GAPDH was used as a loading control. The reaction conditions were first 95°C for 10 min to activate Taq DNA polymerase, followed by denaturation at 95°C for 15 s, and annealing at 60°C for 1 min. The PCR was run 40 cycles, and the final extension cycle was 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s. The fold-change for Cdc6 expression level was calculated using the delta–delta Ct method.
Immunofluorescence
Normal bone marrow mononuclear CML and K562 cells were spun onto polylysine-coated coverslips, washed in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde (PFA) for 5 min at room temperature (RT), permeabilized with PBS including 0.1% Triton X-100 and 0.02% sodium dodecylsulfate (SDS) for 5 min at RT, incubated for 10 min in blocking solution (PBS including 0.1% Triton X-100, 0.02% SDS, and 2% bovine serum albumin (BSA)) to block non-specific adsorption of antibody, incubated with primary antibody in blocking solution for 1 h at 37°C, washed three times, and coverslipped with mounting medium containing 1.5 µg/mL 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories Inc., Burlingame, CA, USA). Images were captured using an Olympus FV500 confocal microscope and were analyzed using Olympus Fluoview v5.0 software.
Western blot analysis
Cells were collected and lysed using mammalian protein extraction reagent (M-PER) lysis buffer (Pierce Biotechnology Inc., Rockford, IL, USA). The protein extracted was quantified using a bicinchoninic acid (BCA) assay kit (Pierce Biotechnology Inc.). An amount of 30 µg proteins of each sample was separated by SDS–polyacrylamide gel electrophoresis (PAGE), transferred to a hydrophobic polyvinylidene difluoride membrane (Hybond-P; Amersham Biosciences, Piscataway, NJ, USA), and immunoblotted with various antibodies according to the manufacturer’s protocol. Chemiluminescence was used to visualize bands according to the manufacturer’s protocol (Amersham Biosciences). Actin was used as a loading control.
siRNA transfection
In separate experiments, an annealed, purified, and desalted double-stranded Cdc6 siRNA (sense 5′-aggcacuugcuaccagcaatt-3′, anti-sense 5′-uugcug guagcaagugccutt-3′); BCR/ABL siRNA (sense 5′-gcagaguucaaaagcccuutt-3′, anti-sense: 5′-aagggcuuuugaacucugctt-3′); and a scramble siRNA against luciferase (sense: 5′-gcgacgaucugccuaagaudtdt-3′, anti-sense: 5′-aucuuaggcagaucgucgcdtdt-3′), obtained from GenePharma Co. Ltd. (Shanghai, China), were applied.
For transfection, 500,000 K562 cells in the exponential phase of growth were seeded in 75 cm2 culture plates and incubated at 37°C for 24 h. SiRNA was complexed with HiPerFect Transfection Reagent (QIAGEN, Hilden, Germany) in Opti-MEM medium (Invitrogen) as described by the supplier. K562 cells were incubated with 200 nmol/L siRNA at 37°C for 6 h and then washed and cultured in RPMI 1640 medium with 10% FBS for 72 and 96 h.
Flow cytometry analysis
K562 cells were treated with Cdc6 siRNA to silence the Cdc6 gene. For cell-cycle analysis, 250,000 cells were cultured at a density of 1 × 106 cells/mL, washed twice with PBS, resuspended in PBS including 0.2% Tween-20 and 0.5 mg/mL Rnase A (Sigma-Aldrich, St. Louis, MO, USA), incubated for 30 min at 37°C, stained with 25.1 µg/mL of propidium iodide (Sigma-Aldrich, St. Louis, MO, USA), and analyzed using a BD FACS Calibur flow cytometer (Becton Dickinson Biosciences, San Jose, CA, USA). The cell-cycle distribution (i.e. relative percentages of cells in the G0/G1 of the cell cycle) was analyzed using ModFit LT for Mac V3.0 software (BD Biosciences).
Apoptotic cells were detected by flow cytometry and measured using an Annexin-V apoptosis assay kit. K562 cells treated by Cdc6 siRNA were collected and double-stained with fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI). For each sample, data from approximately 10,000 cells were collected in list mode and displayed on a logarithmic scale. The percentages of apoptotic cells (PI-negative, Annexin V-positive cells) and necrotic cells (double-positive cells) were analyzed by quadrant statistics.
Cell Counting Kit-8 assay
For cell viability analysis, K562 cells were seeded in 96-well plates at a density of 1 × 106 cells/mL, incubated with reagents, and assayed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Rockville, MD, USA) according to the manufacturer’s instructions. Extinction measured at 540 nm was subtracted from the reference extinction.
Statistical analysis
The data were reported as mean ± standard error of mean (SEM), analyzed using SPSS version 12.0 software, and compared using analysis of variance (ANOVA). Statistical significance was defined as p < 0.05.
Results
Cdc6 was ubiquitously overexpressed in CML cells
To detect the expression of Cdc6, we analyzed normal bone marrow mononuclear cells (BMMCs) from 10 healthy controls, primary CML cells from 10 CML patients, and cell line K562 cells. Normal BMMCs, primary CML, and K562 cells were determined by FISH analysis again. Almost all CML and K562 cells were shown BCR/ABL fusion and a single Ph chromosome (Figure 1(a)). Cdc6 mRNA levels were 10- to 50-fold greater in K562 and primary CML cells than in normal BMMCs (Figure 1(b)). This difference was highly significant (p < 0.05), despite of some variation in Cdc6 expression among CML specimens. To further characterize the de-regulated expression of Cdc6 protein in CML, the expression and cellular distribution of Cdc6 protein were examined in single cells by immunofluorescence staining. Very low level of Cdc6 protein was detectable in cells from healthy controls. In contrast, Cdc6 protein was strongly expressed in most CML cells, exclusively in the nucleus (Figure 2). These data suggested that upregulated expression of Cdc6 at mRNA and protein level may be a common feature in CML cells.

Cdc6 mRNA expression in normal BMMCs (control) and primary CML and K562 cells by quantitative RT-PCR. (a) Primary CML and K562 cells were shown BCR/ABL fusion and single Ph chromosome by FISH analysis. BCR, ABL, and BCR/ABL fusion was labeled with spectrum green, orange, and green and orange, respectively. (b) Cdc6 mRNA expression were upregulated in primary CML and K562 cells by quantitative RT-PCR. Data were reported as mean ± SEM. Each sample was performed in triplicate (n = 10; *p < 0.05 vs control group).

Expression and localization of Cdc6 proteins in normal BMMCs and primary CML cells by immunofluorescence.
Inhibition of BCR/ABL downregulated Cdc6 mRNA expression in K562 cells and primary CML cells in vitro
CML is a hematopoietic malignancy characterized by the presence of chimerical BCR/ABL tyrosine kinase. BCR/ABL can regulate the expression of multiple genes via various mechanisms, having important roles in the initiation and progression of CML disease. So, we wondered whether the ubiquitous overexpression of Cdc6 in CML is related to the tyrosine kinase activity of BCR/ABL. We investigated the effects of the BCR/ABL inhibitor STI571 (0.5, 1, and 2 µmol/L) on Cdc6 expression in K562 cells and primary CML cells using quantitative RT-PCR analysis. STI571 significantly and dose-dependently decreased Cdc6 mRNA expression in K562 cells and primary CML cells (Figure 3(a) and (b)). Our data supported that the constitutive tyrosine kinase activity of BCR/ABL may contribute to the deregulation of Cdc6 expression in CML cells.
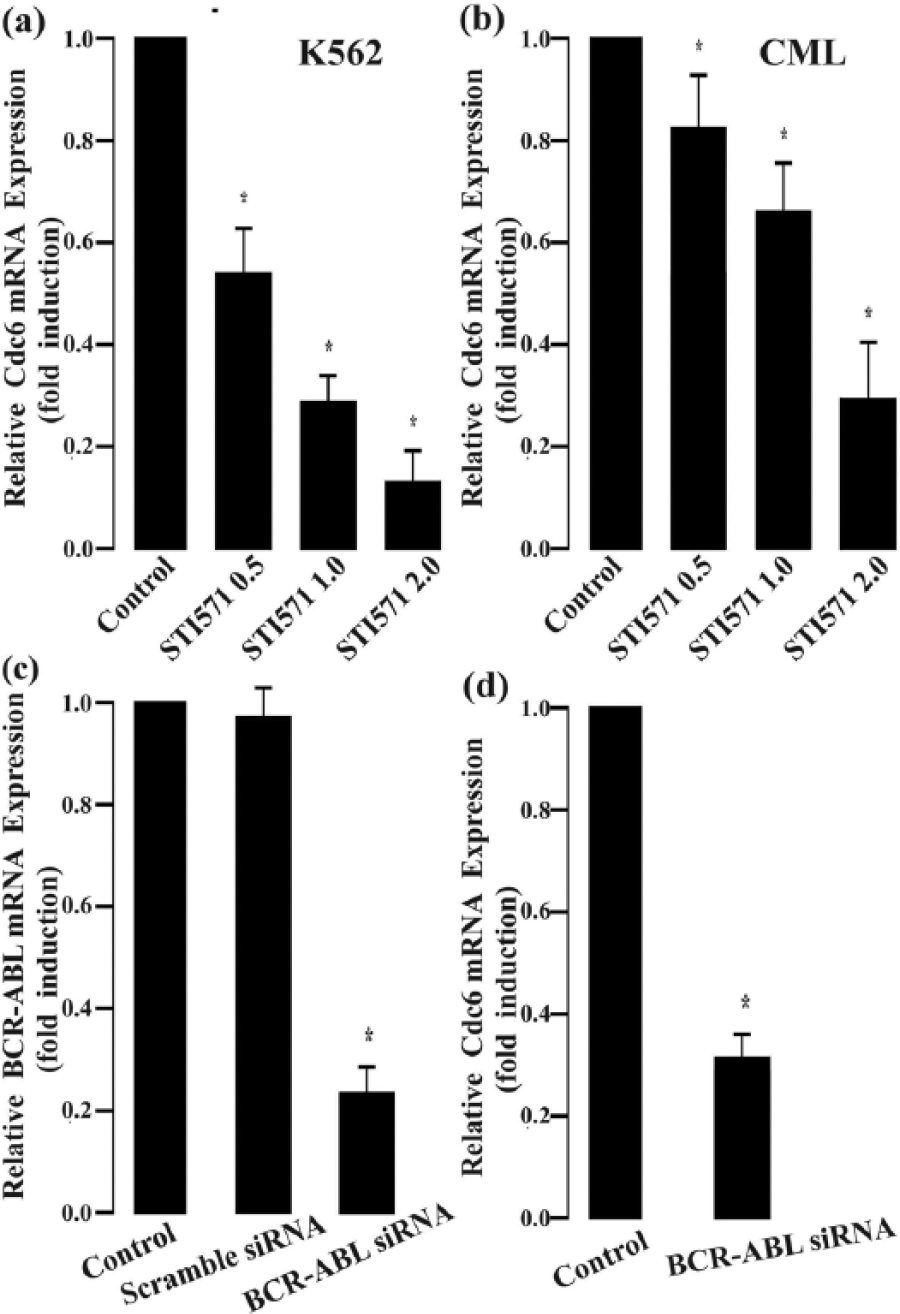
Inhibition of BCR-ABL decreases Cdc6 mRNA expression in primary CML and K562 cells. (a) Cdc6 mRNA expression in K562 cells treated by STI571 (BCR/ABL inhibitor) for 12 h. (b) Cdc6 mRNA expression in primary CML cells treated by STI571 for 12 h. (c) Effects of BCR-ABL gene silencing on K562 cells. K562 cells were tansfected with reagent (control), scramble siRNA, and BCR/ABL siRNA for 72 h, and then, BCR-ABL mRNA expression levels were detected by quantitative RT-PCR. (d) Relative Cdc6 mRNA expression levels in BCR-ABL gene silence K562 cells, and K562 cells were tansfected with scramble siRNA (control) and BCR/ABL siRNA for 72 h, and then, Cdc6 mRNA expression levels were detected by quantitative RT-PCR. Data are reported as mean ± SEM. Each sample was performed in triplicate (n = 5; *p < 0.05 vs control group).
To confirm the role of BCR/ABL in the endogenous Cdc6 overexpression in CML cells, siRNA specific to BCR/ABL was employed to silence BCR/ABL gene in K562 cells, and the resulting effects on Cdc6 expression were examined by real-time quantitative PCR analysis. The results showed that BCR/ABL mRNA expression was decreased by 78% in BCR/ABL siRNA-transfected K562 cells compared to negative control (NC) siRNA-transfected K562 cells (p < 0.001, Figure 3(c)). Consequently, Cdc6 mRNA level was significantly lower in BCR/ABL siRNA-transfected K562 cells than in NC siRNA-transfected K562 cells (p < 0.001, Figure 3(d)). These results further suggested that BCR/ABL may contribute to the deregulation of Cdc6 expression in CML cells.
JAK/STAT and PI3K/AKT pathways were involved in the Cdc6 overexpression in CML cells
JAK/STAT, PI3K/AKT, and ERK signaling pathways are constitutively activated in CML cells due to the presence of chimeric BCR/ABL. STI571 can abolish the activation of these pathways in CML cells. Since STI571 downregulated Cdc6 expression in CML cells, we proposed that constitutive activation of these pathways may be essential to maintain strong Cdc6 expression. To specifically block the activation of JAK/STAT, PI3K/AKT, or ERK signals, K562 cells were treated with AG490 (a JAK/STAT inhibitor), LY294002 (a PI3K/AKT inhibitor), or PD98059 (an ERK inhibitor), respectively. As shown in Figure 4, both LY294002 and AG490 but not PD98059 efficiently decreased Cdc6 mRNA expression in K562 cells, indicating that constitutive activation of the JAK/STAT and PI3K/AKT pathways may contribute to Cdc6 overexpression in CML cells.
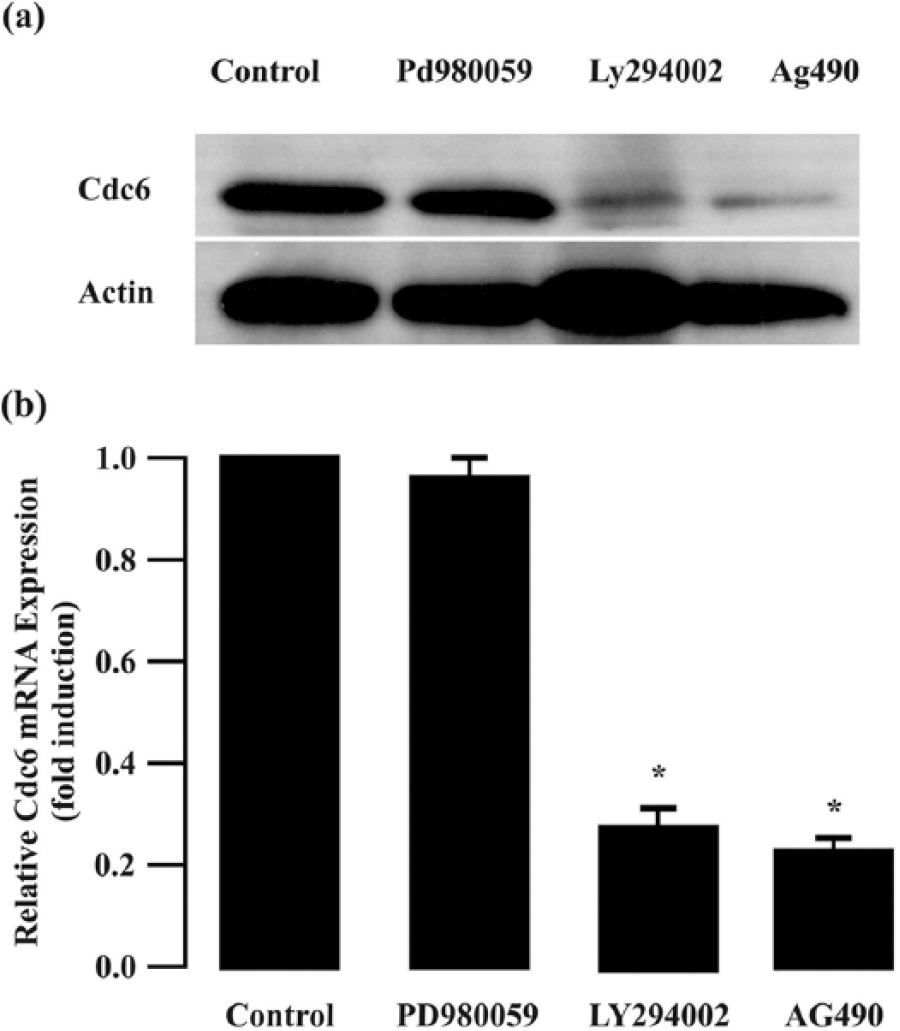
Cdc6 expression in K562 cells treated by PD98059 (an ERK inhibitor), G490 (a JAK/STAT inhibitor), and LY294002 (a PI3K/AKT inhibitor) for 12 h by quantitative RT-PCR and western blot. Data were reported as mean ± SEM. Each sample was performed in triplicate (n = 5; *p < 0.05 vs control group).
Cdc6 gene silencing resulted in decrease in proliferation, increase in apoptosis, and arrest of cell cycle in K562 cells
To further explore the potential roles of endogenous Cdc6 overexpression in CML cells, we observed the effects of Cdc6 gene silencing on the cell proliferation, apoptosis, and cell cycle in K562 cells. K562 cells were transfected with siRNA to specifically knockdown endogenous Cdc6. A scramble siRNA sequence was also transfected into K562 cells as a control; 72 h after transfection, Cdc6 siRNA but not scramble siRNA induced a dramatic reduction in levels of Cdc6 mRNA and protein as determined by quantitative RT-PCR and western blot analysis (Figure 5(a) and (b)).
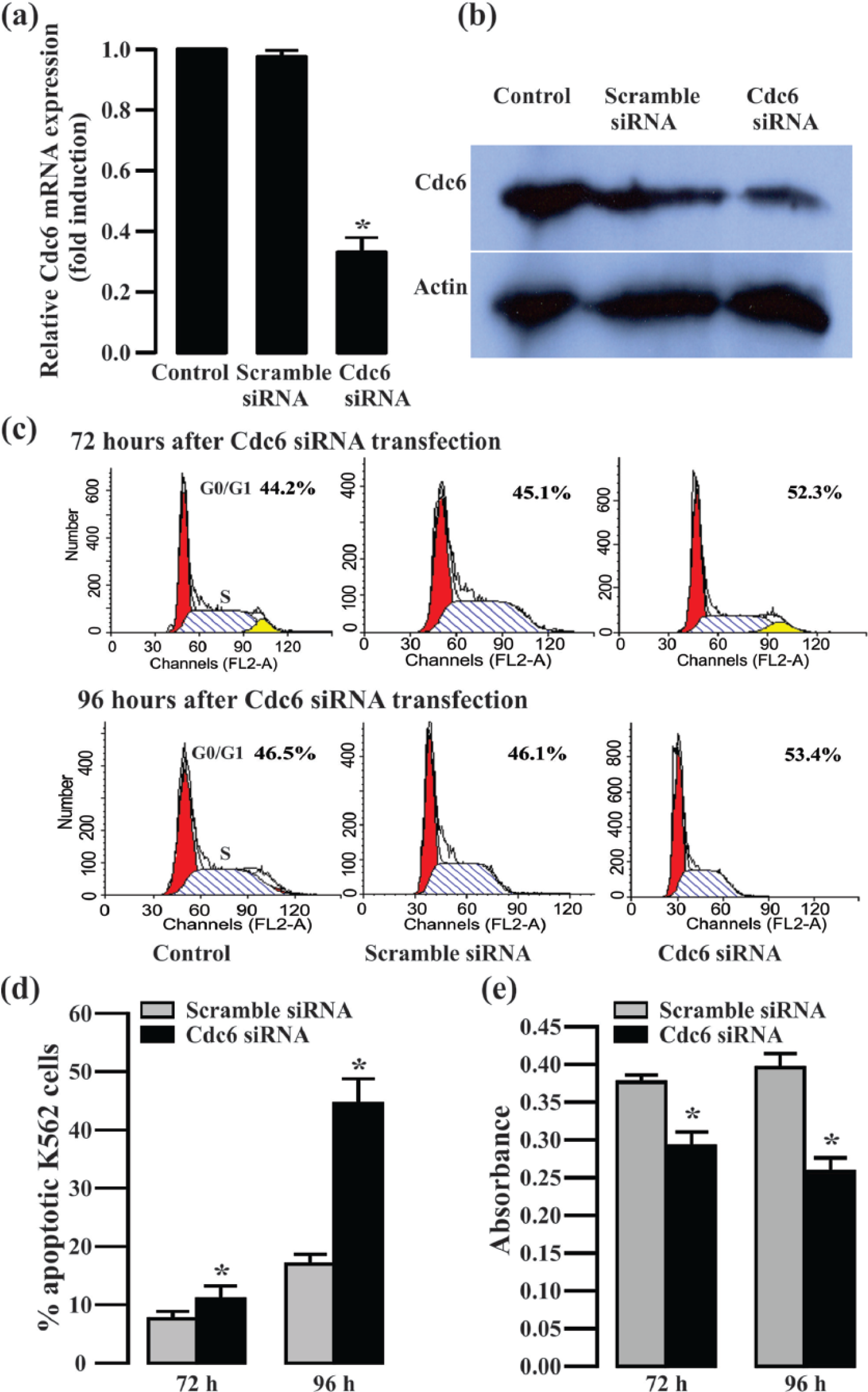
Effects of Cdc6 gene silencing on proliferation, apoptosis, and cell cycle in K562 cells. (a and b) Cdc6 mRNA and protein expression levels were decreased in Cdc6 siRNA–tansfected K562 cells for 72 h by quantitative RT-PCR and western blot. (c) The proportion of cells in G0/G1 phase was increased in Cdc6 siRNA–tansfected K562 cells for 72 and 96 h by flow cytometry analysis. (d) The apoptotic cells were increased in Cdc6 siRNA–tansfected K562 cells for 72 and 96 h by flow cytometry analysis. (e) To study the proliferation effects of K562 cells after Cdc6 gene silencing, the cell viability was decreased in Cdc6 siRNA–tansfected K562 cells for 72 and 96 h by CCK-8 assay. Data were reported as mean ± SEM. Each sample was performed in triplicate (n = 3; *p < 0.05 vs scramble siRNA group).
To study the proliferation effects of K562 cells after Cdc6 gene silencing, cell viability was measured using a CCK-8 kit. Cdc6 gene silencing progressively decreased the viability of K562 cells at 72 and 96 h post-transfection (Figure 5(e)), suggesting that endogenous Cdc6 may promote the growth of CML cells.
Apoptosis was determined by Annexin V-FITC/PI staining. Flow cytometry analysis of K562 cells harvested at 72 and 96 h after Cdc6 siRNA transfection was shown in Figure 5(d). In contrast to the Annexin V-positive fraction of scramble siRNA-transfected K562 cells, the Annexin V-positive fraction of the Cdc6 gene silencing K562 cells was markedly increased at 72 and 96 h after Cdc6 siRNA transfection, indicating significant apoptosis-associated cell death.
Cell-cycle distribution was analyzed by flow cytometry in K562 cells after Cdc6 gene silencing for 72 and 96 h. As shown in Figure 5(c), the proportion of cells in G0/G1 phase was higher in K562 cells transfected with Cdc6 siRNA than in control cells. Moreover, the increase in G0/G1 phase cells was accompanied by a reduction in S-phase cells, suggesting the induction of G1/S arrest.
Discussion
Recent evidence has suggested that the abnormal Cdc6 expression is closely associated with oncogenic activity in some human cancers.21–23 Though regulatory mechanisms normally maintain Cdc6 protein levels close to physiological levels, these levels can be abnormally high in some human tumors. 24 Overexpressed Cdc6 usually promotes tumor development by negatively regulating tumor suppressor genes. Therefore, altering the abnormal expression of Cdc6 might be a valuable strategy for cancer treatment.25,26 It would be of great interest to understand whether Cdc6 expression affects human CML cells. In this study, Cdc6 was deregulated/overexpressed in the human CML cell line K562 and primary CML cells. Moreover, Cdc6 protein was found exclusively in the nuclei of normal and CML BMMCs by immunofluorescence. Obviously, overexpression of Cdc6 may play an important role in the pathogenesis of CML.
Previous studies have linked inhibition of Cdc6 expression via Cdc6 knockdown with suppressed proliferation of some human neuroblastoma cells.27,28 However, the involvement of Cdc6 in CML has rarely been characterized and studied. In this study, Cdc6 controlled CML cell proliferation and death. Gene silencing of Cdc6 expression via siRNA suppressed CML cell proliferation, induced CML cell apoptosis, and caused the accumulation of G0/G1 populations along with the depletion of the S-phase population. In addition, some studies have shown that high levels of Cdc6 do not necessarily correlate with increased tumor cell proliferation. 12 In the case of NSCLCs, the level of Cdc6 did not directly correlate with the level of proliferation marker Ki67. 12 However, our results showed that Cdc6 gene silencing suppressed cell proliferation and increases cell death, thereby reducing tumor cell malignancy. These data indicated a close association of Cdc6 with the pathogenesis of CML.
We therefore were interested to know whether the disease-related oncogene BCR/ABL plays a role in the expression of Cdc6 in leukemic cells. First, we found that BCR/ABL inhibitor STI571 downregulated expression of Cdc6 in CML cells. Some studies have found that BCR/ABL activates PI3K/Akt, Ras/Raf/MEK/ERK, and JAK/STAT signal transduction pathways during the progression of CML.20,29,30 In our study, specific inhibitors of the PI3K/Akt and JAK/STAT pathways (LY294002 and AG490, respectively) significantly reduced Cdc6 expression. Collectively, these results suggest that activation of PI3K/Akt and JAK/STAT pathways may play an essential role in mediating the regulatory effect of BCR/ABL on Cdc6 expression in CML cells. How they downregulate Cdc6 expression remains unclear and merits further investigation.
In conclusion, Cdc6 overexpression is involved in oncogenic activity in human CML. Specifically, gene silencing of Cdc6 is associated with human CML cell proliferation and apoptosis. Also, inhibiting BCR/ABL tyrosine kinase activity can downregulate Cdc6 expression. Thus, Cdc6 protein may be regulated by BCR/ABL signal transduction through downstream PI3K/Akt and JAK/STAT pathways in CML cells and an attractive target for the development of effective anti-cancer strategies in CML.
Footnotes
Acknowledgements
J.-H.Z., Y.-L.H., and R.Z. have contributed equally to this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (No. 81371102) and the Fundamental Research Funds for the Central Universities (No. 2015QN210).