
Abstract
Aurora kinases play critical roles in regulating several processes pivotal for mitosis. Radotinib, which is approved in South Korea as a second-line treatment for chronic myeloid leukemia, inhibits the tyrosine kinase BCR-ABL and platelet-derived growth factor receptor. However, the effects of radotinib on Aurora kinase expression in acute myeloid leukemia are not well studied. Interestingly, the cytotoxicity of acute myeloid leukemia cells was increased by radotinib treatment. Radotinib significantly decreased the expression of cyclin-dependent kinase 1 and cyclin B1, the key regulators of G2/M phase, and inhibited the expression of Aurora kinase A and Aurora kinase B in acute myeloid leukemia cells. In addition, radotinib decreased the expression and binding between p-Aurora kinase A and TPX2, which are required for spindle assembly. Furthermore, it reduced Aurora kinase A and polo-like kinase 1 phosphorylation and suppressed the expression of α-, β-, and γ-tubulin in acute myeloid leukemia cells. Furthermore, radotinib significantly suppressed the key regulators of G2/M phase including cyclin B1 and Aurora kinase A in a xenograft animal model. Therefore, our results suggest that radotinib can abrogate acute myeloid leukemia cell growth both in vitro and in vivo and may serve as a candidate agent or a chemosensitizer for treating acute myeloid leukemia.
Introduction
Aurora kinases (AURKs) are a family of three highly homologous serine/threonine kinases that play critical roles in regulating processes that are crucial for mitosis.1,2 Different AURK isoforms, namely, Aurora kinase A (AURKA), Aurora kinase B (AURKB), and Aurora kinase C, play various regulatory functions in mitosis. AURKA is involved in regulating several early mitotic events, including entry into mitosis. 3 The levels of AURKA increase during the G2 phase and peak early in mitosis. 4 Cdc25B, a direct regulator of the cyclin B1–cyclin-dependent kinase 1 (CDK1) complex, is an AURKA target, which underscores the function of this enzyme in regulating mitotic entry. AURKA also regulates centrosome maturation by moderating the recruitment of proteins, such as TPX-2 and Bora, which are themselves essential for assembly of microtubule spindle components, namely, γ-tubulin.3,5 In addition, AURKA is associated with centrosome separation. 1
Since their discovery in the 1990s, AURKs have been associated with the progression of human cancers. 6 AURKA and AURKB are mapped to 20q13 and 17q13.1 on human chromosomes, respectively, which are loci frequently altered in human cancers. 7 Overexpression of AURKA and AURKB is seen in many cancers, including colon, breast, ovarian, stomach, and pancreatic cancers.1,6,8,9 Therefore, inhibition of AURKs forms a key therapeutic approach for a broad range of cancers.2,10 Indeed, AURK inhibitors are being actively studied for the development of anticancer therapies. Current preclinical data regarding AURK inhibition highlight the utility of this approach for the treatment of several cancers. 2
Acute myeloid leukemia (AML) is one of the most difficult hematological malignancies to cure.11,12 The current standard therapy for AML is based on old chemotherapeutic regimens, which leaves room for considerable improvements. Many clinicians consider AURK as a good therapeutic target for AML, which is supported by clinical evidence. Recently, AURK inhibitors, including AT9282, MLN-8237, and KW-2449, were evaluated in clinical trials for their efficacy in treating leukemia, where they showed promising activity in AML and Philadelphia chromosome-positive leukemia. 13 Moreover, recent advances in the development of AURK inhibitors as therapeutics for hematological malignancies have attracted considerable attention.13–15
Radotinib is approved in South Korea as a second-line treatment for chronic phase chronic myeloid leukemia (CML-CP).16,17 It inhibits the tyrosine kinase BCR-ABL and platelet-derived growth factor receptor.16,18 Recently, Phase III clinical trial on the efficacy and safety of radotinib demonstrated its superiority over imatinib in generating complete cytogenetic response and major molecular response in patients newly evaluated with Philadelphia chromosome-positive CML-CP. 19 Radotinib showed neuroprotective effects in a preclinical Parkinson’s disease mouse model. 20 Besides, it effectively accesses the brain and exhibits greater pharmacokinetic properties and safety profiles compared to nilotinib and other c-Abl inhibitors. More recently, radotinib activated natural killer cell cytotoxicity against various Fas-expressing solid cancer cells, including lung, breast, and melanoma cells. 21 Previously, we presented that radotinib increased cytotoxicity of all types of AML cells.22,23 It inhibited AML cell proliferation by activating mitochondria-dependent apoptosis and inducing the expression of CDK inhibitors. 22 Moreover, radotinib induced apoptosis in CD11b+ cells differentiated from AML blast cells. 23 Furthermore, targeting of c-KIT (CD117) by radotinib promoted AML cell death in c-KIT-positive AML cells.24,25 However, the effects of radotinib on AURK expression in AML cells are not well known and further studies on the mechanism of radotinib-mediated cell cycle arrest are required. In this study, we demonstrate antileukemic effects of radotinib by inhibition of mitosis and suppression of AURKA expression in AML cells.
Materials and methods
Reagents
Radotinib was a generous gift from Ilyang Pharmaceutical Co. Ltd. (Seoul, South Korea) and was found to be 99.9% pure after high-performance liquid chromatography (HPLC) analysis. 23 All reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise indicated. Apoptosis detection kit I and propidium iodide (PI)/RNase solution were purchased from BD Biosciences (San Jose, CA, USA). The CellTiter 96 AQueous One Solution cell proliferation assay system was purchased from Promega (Madison, WI, USA). All antibodies for Western blotting were purchased from Cell Signaling Technology (Beverly, MA, USA), and antibody for immunoprecipitation against TPX2 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Ethics statement
All mice experiments were performed in accordance with the relevant guidelines and regulations established by the ethical guidelines and regulations of the Korean Association for Laboratory Animals, and all experimental procedures were approved by the Institutional Animal Care and Use Committee of the Ulsan University of Korea (approval no.: 0117-07).
Cell culture
The human AML cell lines in this study were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and they were grown as suspension cultures in 100-mm culture dishes in RPMI-1640 medium with 10% fetal bovine serum (FBS) (or 20% FBS for the Kasumi-1 cells) and 1% penicillin–streptomycin solution in a 5% CO2-humidified atmosphere at 37°C, as described previously. 23
Cell viability assay
Kasumi-1 cells were incubated with 0, 1, 2, and 5 µM radotinib for 48 h at 37°C, followed by harvesting and washing twice with phosphate buffered saline (PBS). The MTS assay was performed using the CellTiter 96 solution, as described previously,22,23 and cell viability was assessed using the Muse® viability assay kit (Merck Millipore), according to the manufacturer’s instructions. Samples were analyzed using Muse® cell analyzer and its associated software.
Cell proliferation assay (BrdU assay)
Kasumi-1 cells were incubated with 0, 1, and 5 μM radotinib for 48 h at 37°C. Cell proliferation was measured by 5′-bromo-2-deoxyuridine (BrdU) enzyme-linked immunosorbent assay (Cell Proliferation ELISA, BrdU; Roche Diagnostics, IN, USA), according to the manufacturer’s instructions. The results are expressed as percentage changes from the basal condition using three to five culture wells for each experimental condition.
Cell cycle analysis
Kasumi-1 cells (5 × 105 cells/mL) were seeded in 24-well plates and treated with 0, 1, and 5 μM radotinib for 48 h at 37°C. They were washed twice with PBS and fixed with 70% ethanol overnight at −20°C, followed by washing again with PBS and incubation with 0.5 mL PI/RNase stain buffer for 15 min at room temperature. The samples were then analyzed using a FACSCalibur flow cytometer and CellQuest Pro software (BD Biosciences).
Intracellular staining of AURKA
Kasumi-1 cells were incubated with 0, 1, and 5 μM radotinib for 48 h at 37°C, followed by harvesting and washing twice with FACS buffer (PBS containing 0.1% NaN3 and 0.2% bovine serum albumin). Next, they were fixed with 4% paraformaldehyde in PBS, after which they were added to a solution of 0.1% Triton X-100 in PBS for permeabilization. The cells were stained with anti-AURKA monoclonal antibody or isotype control monoclonal antibody at 4°C for 30 min. The samples were then analyzed using a FACSCalibur flow cytometer and CellQuest Pro software.
Microarray analysis
Kasumi-1 cells were incubated with 0 and 5 µM radotinib for 48 h and were analyzed using a 44K oligo-microarray (Agilent Technologies, Inc., Palo Alto, CA, USA). Microarray analysis was performed as described previously. 23
Microarray data analysis
Microarray data were analyzed as described previously. 23 Expression changes of >2-fold were considered significant. For understanding expression patterns, hierarchical clustering analysis was performed using the GeneSpring software. Functional enrichment analyses were performed using the Gene Ontology (GO) functional classification system (www.geneontology.org) or DAVID (http://david.abcc.ncifcrf.gov/) (GEO accession: GSE72926).
Western blot analysis and immunoprecipitation
The cells were lysed as described previously for immunoprecipitation. 23 Kasumi-1 cells (1 × 107 cells/mL) were incubated with radotinib for 48 h and lysed in immunoprecipitation assay buffer for 30 min at 4°C. Equal amounts of proteins were precleared with protein G beads. The beads were pelleted, and the supernatant was incubated overnight with appropriate antibodies and beads. Equal quantities of solubilized protein were resolved by 10%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) as described previously. 23
Xenograft animal model using Kasumi-1 cells
Specific pathogen-free 5-week-old athymic nude male mice were purchased from Koatech (Pyeongtaek, Korea) and maintained in a clean environment. The mice received whole body irradiation with 6-MV photon at a dose of 4 Gy. At 16–24 h postirradiation, the mice were subcutaneously implanted with Kasumi-1 cells (3 × 107 cells/mouse with 0.2 mL of matrigel) in the right flank region. The mice were randomly assigned to three groups based on the radotinib concentration used (0, 10, and 20 mg/kg, n = 4). When the tumors were ∼150 mm3 in size at ∼7 days postimplantation, 0.2 mL radotinib (0, 10, and 20 mg/kg body weight) was injected intraperitoneally five times per week on different days. The tumor’s maximal length and width were measured once a week using a digital caliper, and the tumor volume (V) was calculated using the following formula: V = (length × width)2 × 0.5. The mice were sacrificed on days 30–32 following tumor cell implantation. The body weight of the tumor-bearing mice did not change significantly during the duration of the study. The tumors were excised and weighed, and each tumor tissue was homogenized for the preparation of cell samples for several analyses.
Statistics
The data presented here represent mean ± standard error of mean (SEM) of at least three independent experiments. All values were evaluated using one-way analysis of variance followed by Tukey’s range test in GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, USA). Differences were considered significant at p < 0.05. Each treatment was assayed in triplicate.
Results
Radotinib induces cell death of Kasumi-1 cells
According to our results, radotinib induced cell death in different types of AML cells. Among them, Kasumi-1 cells were the most sensitive to radotinib, and it is well known that the Kasumi-1 cell line is good for an AML of the t(8;21)-kit mutant model. 26 For this reason, experiments on Kasumi-1 cells were conducted in this study. We tested diverse effects of radotinib in Kasumi-1 cells. Radotinib decreased cell viability, proliferation, G2/M phase, and the levels of survival proteins such as BCL-xL and BCL-2 and G2/M phase-related proteins (CDK1 and cyclin B1) in a dose-dependent manner (Figure 1(a)–(i)). It also increased the number of dead and apoptotic cells (Figures 1(c) and (e)). Thus, these results indicate that radotinib induces cell death of AML cells, especially Kasumi-1 cells.

Effect of radotinib on Kasumi-1 cells. Cells were incubated with 5 μM radotinib for 48 h, harvested, and used for multiple experiments. (a) Live and dead cells were analyzed using the Muse® viability assay kit and Muse cell analyzer. (b) Average number of live cells. (c) Average number of dead cells. (d) Viability of Kasumi-1 cells. (e) Apoptotic cells were monitored by annexin V staining. (f) Expression analysis of BCL-xL and BCL-2 by Western blotting. (g) Cell proliferation was monitored by BrdU staining. (h) Cell cycle distribution was analyzed by PI/RNAse staining. (i) The levels of CDK1 and cyclin B1 were determined by Western blotting. β-Actin was used to confirm equal loading.
Radotinib suppresses the expression of AURKA and AURKB in Kasumi-1 cells
Microarray results showed that radotinib treatment of Kasumi-1 cells led to >2-fold differences in the expression of many genes related to apoptosis and the cell cycle. Radotinib-responsive genes by GO categories are shown in Table 1. These results indicate that radotinib simultaneously induces apoptosis and inhibits proliferation and cell cycle. Interestingly, radotinib significantly inhibited the expression of AURKA and AURKB (both mRNA and protein) in Kasumi-1 cells (Figure 2(a)–(f)).
Radotinib-responsive genes by GO categories.

GO: Gene Ontology; MAP: mitogen-activated protein; MAPKKK: mitogen-activated protein kinase kinase kinase.
GO analysis was performed by DAVID Bioinformatics Resources. Shown are GO Biological process terms that significantly overrepresented for the radotinib-responsive genes (p value < 0.01).

Radotinib suppresses the expression of AURKA and AURKB in Kasumi-1 cells. (a) Gene expression by microarray. Kasumi-1 cells were incubated with 5 μM radotinib for 48 h. Cells were harvested, total RNA was isolated, and subjected to microarray analysis as described in section “Materials and methods.” (b) Protein levels of AURKA and AURKB after treatment of Kasumi-1 cells with radotinib (0, 0.5, 1, and 5 μM). β-Actin was used to confirm equal loading. (c, d) Relative band density of AURKA and AURKB shown in (b). (e) Intracellular staining of AURKA under the same condition. Green histogram indicates AURKA in cells treated with 0 μM radotinib, and black histogram indicates AURKA in cells treated with 5 μM radotinib. (f) Positive cells of intracellular AURKA shown in (e).
Radotinib inhibits AURKA activity on substrate proteins in Kasumi-1 cells
Radotinib significantly reduced AURKA expression (Figure 2(a)–(c), (e), and (f)). Therefore, we investigated its effect on AURKA substrates. Generally, polo-like kinase 1 (PLK1), which is activated by AURKA upon phosphorylation at the G2/M transition, plays important roles during mitosis. 3 As shown in Figure 3(a), active PLK1 is involved in the promotion of mitotic entry via activation of cdc25, CDK1, and cyclin B1.1,3 Radotinib inhibits both expression and activity of AURKA in Kasumi-1 cells (Figure 3(b)). In addition, PLK1 (Thr210) activity was completely inhibited via radotinib-induced suppression of AURKA expression (Figure 3(c)). Moreover, the expression of PLK1, Bora, cdc25A, and cdc25B of the PLK axis was significantly abolished by radotinib in Kasumi-1 cells (Figure 3(c)).

Radotinib inhibits AURKA activity in Kasumi-1 cells. Cells were incubated with radotinib for 48 h under the same condition as mentioned in Figure 2. Cells were harvested and protein levels were monitored by Western blotting. (a) Diagram showing the role of AURKA in mitosis. (b) The levels of AURKA and p-AURKA after radotinib treatment. (c) p-PLK1, PLK1, Bora, cdc25A, and cdc25B expression after radotinib treatment. β-Actin was used to confirm equal loading.
Radotinib decreases the binding between p-AURKA and TPX2 for spindle assembly and the expression of α-, β-, and γ-tubulin
Radotinib decreased the expression of and binding between p-AURKA and TPX2, which is required for spindle assembly (Figure 4(a)). As a result, the levels of α-, β-, and γ-tubulin were completely reduced in Kasumi-1 cells (Figure 4(b) and (c)).

Radotinib decreases the binding between p-AURKA and TPX2, which is required for spindle assembly, and the expression of α-, β-, and γ-tubulin. Kasumi-1 cells were incubated with radotinib for 48 h under the same conditions as mentioned in Figure 2. (a) Cells were harvested and the binding of p-AURKA to TPX2 was observed by immunoprecipitation. (b) The expression of α-, β-, and γ-tubulin was monitored by Western blotting. (c) Intracellular staining of α-, β-, and γ-tubulin. β-Actin was used to confirm equal loading.
Responses of Kasumi-1 cells to radotinib and the pan-AURK inhibitor, VX-680, are similar
To determine the mechanism underlying the effects of radotinib and understand the role of AURK in inducing cell death and CD11b expression in Kasumi-1 cells, we treated cells with the pan-AURK inhibitor, VX-680 (MK-0457, Tozasertib; 0, 10, 30, and 50 nM). 27 Radotinib decreased cell viability in a dose-dependent manner (Figure 5(a)) and increased the number of dead and apoptotic cells (Figure 1(c) and (e)). As shown in Figure 2, the levels of AURKA and AURKB were markedly reduced by radotinib. Moreover, radotinib also increased the expression of CD11b, a myeloid-associated differentiation marker, in Kasumi-1 cells (Figure 5(c)). The responses of Kasimu-1 cells to radotinib are similar to those elicited after treatment with the pan-AURK inhibitor, VX-680. Briefly, VX-680 significantly reduced cell viability and the expression of α-, β-, and γ-tubulin, CDK1, and cyclin B1 in Kasumi-1 cells (Figure 5(b), (e), and (f)). VX-680-mediated stimulation of CD11b expression in Kasumi-1 cells was similar to that observed after radotinib treatment (Figure 5(d)). Therefore, these results suggest that radotinib functions as an AURK inhibitor in AML cells (Figure 5).
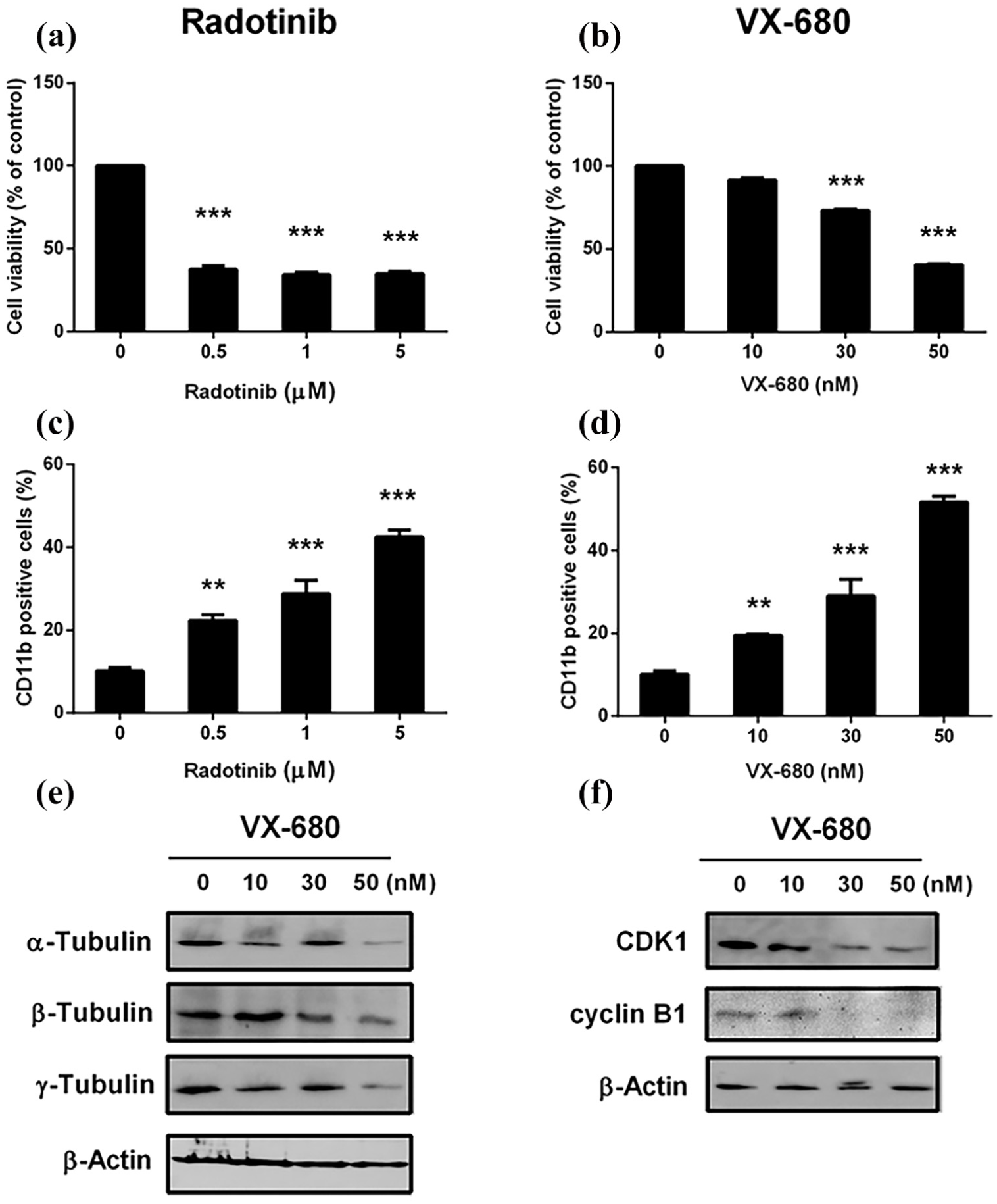
Response of Kasumi-1 cells to radotinib and the pan-AURK inhibitor, VX-680, is similar. Cells were incubated with 0, 0.5, 1, and 5 μM radotinib and 0, 10, 30, and 50 nM VX-680 for 48 h, respectively. (a) Cell viability after radotinib treatment was determined by the MTS assay. (b) Cell viability after VX-680 treatment. (c and d) CD11b expression after radotinib and VX-680 treatments. (e) α-, β-, and γ-tubulin protein expression after VX-680 treatment. (f) CDK1 and cyclin B1 expression after VX-680 treatment. β-Actin was used to confirm equal loading.
Radotinib inhibits the expression of AURKA, CDK1, cyclin B1, and α-, β-, and γ-tubulin in various AML cell lines
In addition, we evaluated the effects of radotinib on AURKA, CDK1, cyclin B1, and α-, β-, and γ-tubulin expression in various AML cell lines, including HL60, HEL92.1.7, and THP-1 under the same conditions as mentioned earlier. We observed that radotinib decreased the expression of these proteins (Supplementary Figure 1(a)–(c)). The reduction in AURKA, CDK1, and cyclin B1 levels in AML cells leads to G0/G1 phase arrest, possibly because of the radotinib-mediated regulation of the CDK inhibitor (CDKI)–CDK–cyclin cascade in AML cells (Supplementary Figure 1). Furthermore, the decrease in the expression of α-, β-, and γ-tubulin might affect spindle assembly in AML cells.
Radotinib inhibits AML cell growth in vivo
To evaluate the role of radotinib in AML cell growth in vivo, we engrafted Kasumi-1 cells in nude mice. Radotinib significantly suppressed tumor growth and reduced tumor volume and weight in a dose-dependent manner (Figure 6(a)–(c)). The body weight of the tumor-bearing mice did not change significantly during the duration of the study. In addition, radotinib inhibited expression of cyclin B1, BCL-xL, BCL-2, AURKA, and α- and γ-tubulin in Kasumi-1 cells isolated from the tumor tissue (Figure 6(d)). Our results showed that radotinib significantly inhibited AML cell growth in vivo and may be potentially used for anti-AML therapy in future.

Radotinib inhibits tumor growth in the xenograft animal model (n = 4 for each group). (a) Representative photographs of tumors 24 days after radotinib treatment. Radotinib (0.2 mL; 0, 10, and 20 mg/kg body weight) was injected intraperitoneally five times per week on different days when the tumors were ~150 mm3 at ~7 days postimplantation. (b) Determination of tumor volume (mm3). Tumor sizes were measured once a week using a digital caliper, and tumor volumes were calculated using the formula: (length × width)2× 0.5. (c) Determination of tumor weight (g). (d and e) The expression of AURKA, cyclin B1, BCL-xL, BCL-2, AURKA, and α- and γ-tubulin in Kasumi-1 cells isolated from the tumor tissue. β-Actin was used to confirm equal loading.
Discussion
One of the tyrosine kinase inhibitors (TKIs), dasatinib, has been developed to treat CML as a multitargeted TKI, but recent studies show that the application of AML therapy is progressing strongly in both basic and clinical studies. Johnson group in Pittsburgh presented that dasatinib promoted all-trans retinoic acid (ATRA)-induced differentiation of AML cells. 28 In general, acute promyelocytic leukemia (APL), a subtype of AML, undergoes differentiation in response to treatment with ATRA, and the combination of ATRA and chemotherapy shows a high rate of treatment in this disease. But some patients can develop resistance to the treatment. Therefore, alternative or combination therapies could be necessary to improve prognosis and survival. The combination of dasatinib and ATRA showed a definite therapeutic effect through the differentiation of APL cells. Moreover, recent Phase 1 clinical studies have been published reflecting these baseline results. 29 Furthermore, signaling mechanism of dasatinib on AML cell differentiation was related to MEK/ERK-dependent activation of STAT1 and association of Lyn and c-Raf.30,31 Also, dasatinib showed various studies on the therapeutic potential of the t(8;21)-kit mutant and core-binding factor (CBF) AML model.24,26,32 Moreover, c-KIT (CD117) targeting by dasatinib promoted AML cell death. 24 Based on these basic results, dasatinib was added to intensive induction and consolidation chemotherapy and administered as a single agent for 1-year maintenance in first-line treatment of CBF AML (results of the AMLSG 11-08 trial). 33 In addition, the results of combined therapy with dasatinib and other inhibitors including FLT3 inhibitors, JAK inhibitors, and BCL-2 inhibitor (navitoclax) showed high synergism.34,35
Another TKI, radotinib have also been developed to treat CML, and studies on the application of AML therapy are similar to those of dasatinib. Interestingly, our previous studies showed that radotinib induced cytotoxicity in AML cells of diverse karyotypes.22,23 In brief, we compared the effects of radotinib and dasatinib (as well-known TKI) on AML cell viability. The effects of radotinib are very similar to those of dasatinib on diverse AML cell lines and small cell lung cancer cell line (data not shown). In previous studies, radotinib inhibited AML cell proliferation by activating mitochondria and caspase-dependent apoptosis. 22 In addition, it induced G0/G1 phase arrest by inducing CDKIs p21 and p27 and by inhibiting CDK2, CDK4, and CDK6. We have shown that the results about targeting CDK2, 4, and 6 by radotinib promote AML cell death. These results suggested that radotinib functioned as a CDK inhibitor in AML cells. 22 Moreover, targeting of c-KIT (CD117) by radotinib promoted AML cell death in c-KIT-positive AML cells.24,25 We have already shown the results about the effects of radotinib on c-KIT-positive AML cells, and the evidence of specificity for c-KIT expression using cell sorter. 25 Therefore, we realized that radotinib induced high cytotoxicity in c-KIT AML cells. According to our previous results, the c-KIT targeting with radotinib promotes AML cell death, and c-KIT endocytosis is essential for triggering c-KIT-positive AML cell death by radotinib during the early stages. In addition, radotinib reduces heat shock protein 90β (HSP90β) expression (both mRNA and protein) and releases Apaf-1 in c-KIT-positive AML cells. Finally, reduction of the HSP90β expression by radotinib treatment was shown to accelerate AML cell death. Therefore, these results proposed that radotinib functioned as an HSP90 inhibitor in AML cells.
Previously, we validated that radotinib caused G0/G1-phase cell cycle arrest and reduced the proliferation rate of AML cells. 22 However, the signaling pathways involved in the G0/G1-phase cell cycle arrest by radotinib on AML cells are not well known. Moreover, little is known about the effect of radotinib on the expression of AURK in AML cells, and an in-depth study of the cell cycle arrest mechanism by radotinib is needed. In this study, we demonstrate the effects of radotinib on the signaling molecules that are influenced and/or targeted including AURKA in AML cells and their underlying mechanism.
Radotinib decreased the expression of CDK1 and cyclin B1, the key regulators of G2/M phase (Figure 1). Moreover, it inhibited the expression of AURKA and AURKB in AML cells (as shown by the results of microarray and Western blotting; Figure 2). In addition, radotinib reduced the binding between p-AURKA and TPX2 (Figure 4), inhibited the expression of α-, β-, and γ-tubulin and decreased PLK1 phosphorylation in AML cells (Figures 3 and 4). These results indicate that radotinib-mediated reduction in AURKA levels inhibits mitosis entry and tubulin expression in AML cells (Figures 2 –4). Moreover, the response of Kasumi-1 cells to radotinib was similar to those elicited after treatment with VX-680, a known pan-AURK inhibitor (Figure 5). Therefore, radotinib functions as an AURK inhibitor in AML cells. In addition, it repressed the expression of AURKA, CDK1, cyclin B1, and α-, β-, and γ-tubulin in various AML cell lines such as HL60, HEL92.1.7, and THP-1 (Supplementary Figure 1). Furthermore, radotinib significantly inhibited AML cell growth in vivo in a xenograft model (Figure 6). More interestingly, radotinib inhibited cyclin B1, BCL-xL, BCL-2, AURKA, and α- and γ-tubulin expression in Kasumi-1 cells isolated from the tumor tissue (Figure 6(d)). Thus, radotinib inhibits mitosis entry in AML cells via suppression of AURKA expression. These results suggest that radotinib functions as an AURK inhibitor in AML cells.
AURKA is an essential molecule for cell cycle progression and is associated with G2/M transition.1,3 Hence, AURKs have been considered new targets for cancer therapy,1,2,36 and AURK inhibitors have attracted attention for the treatment of different types of cancers, including solid tumors and hematological malignancies.7,10,13,37–39 According to recent research trends, Qi et al. 40 presented that AT9283, a novel AURK inhibitor, suppresses tumor growth in aggressive B-cell lymphomas. Another AURK inhibitor, MLN8237, in combination with docetaxel enhances apoptosis and antitumor activity in mantle cell lymphoma. 41 These results suggest that Aurora inhibitor may represent a novel therapeutic strategy that could be evaluated in relapsed or refractory aggressive B-cell non-Hodgkin lymphomas. Also, Gorgun et al. 42 showed that AURKA inhibitor, MLN8237, induces cytotoxicity and cell cycle arrest in multiple myeloma cells. Therefore, treatment with Aurora inhibitor is considered to be an important treatment for hematologic malignancies and is being developed as one of the indispensable treatment methods for hematologic malignancies based on various basic studies. In this regard, it is interesting that radotinib can act as an AURK inhibitor. In addition, our results will provide information that currently well-known TKIs can act as AURK inhibitors.
It is very difficult to set a xenograft animal model with Kasumi-1 cells. There are also very few known references. But Zhang et al. 43 showed a good protocol very well. We followed and modified the protocol and got good results. The reason why the animals were treated with 4 Gy radiation before the AML cells were implanted was to help to grow well the Kasumi-1 cells that were implanted in mice body. In brief, the immune system of mice could suppress by 4 Gy radiation. So, treatment of 4 Gy radiation is a very useful protocol or process to make the xenograft animal model (especially, using Kasumi-1 cells).
In conclusion, radotinib accelerated G0/G1-phase cell cycle arrest by inhibiting AURKA expression, which inhibited mitosis entry in AML cells. Therefore, our results indicate that radotinib can abrogate AML cell growth (or proliferation) both in vitro and in vivo and may serve as a candidate agent or a chemosensitizer for treating AML. In addition, radotinib might be used in combination with other chemotherapeutic agents such as conventional chemotherapies (cytarabine and anthracyclines), hypomethylating agents (5-azacytidine (5-AZA) and decitabine), and FLT3 inhibitors for AML treatments. This study provides the first evidence that radotinib could be used as an effective therapeutic to treat AML via suppression of AURKA expression, suggesting that radotinib treatment has a potential role in antileukemic therapy on AML. In particular, no reports have shown any association between AURKA and TKI including dasatinib and radotinib.
Supplemental Material
Supplementary_Information_20190124 – Supplemental material for Radotinib inhibits mitosis entry in acute myeloid leukemia cells via suppression of Aurora kinase A expression
Supplemental material, Supplementary_Information_20190124 for Radotinib inhibits mitosis entry in acute myeloid leukemia cells via suppression of Aurora kinase A expression by Sook-Kyoung Heo, Eui-Kyu Noh, Yoo Kyung Jeong, Lan Jeong Ju, Jun Young Sung, Ho-Min Yu, Jaekyung Cheon, SuJin Koh, Young Joo Min, Yunsuk Choi and Jae-Cheol Jo in Tumor Biology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2017R1A1A3A04069314, 2017); the Biomedical Research Center, funded by the Ulsan University Hospital (UUHBRC-2016-001); and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI17C0904).
Supplemental material
Supplementary information includes one figure and can be found in the online version of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.