
Abstract
Differentiation therapy is directed to the self-renewing cancer stem cells, as well as their progeny transit amplifying cells, to force them to mature to terminal differentiation. Differentiation therapy is effective in treatment of neuroblastomas and myeloid leukemias. Checkpoint inhibition therapy removes blocks to cancer reactive T-killer cells and allows them to react to malignant cells and limit the growth of cancer. The percentage of patients with a given cancer that responds to either therapy is less than hoped for, and the duration of response is variable. Multiplying the response rate (percentage of patients responding to therapy) by the duration of response may be used to derive a survival score for patients treated with differentiation therapy or checkpoint inhibition. By this criterion, differentiation therapy gives better survival scores than checkpoint inhibition. Yet, checkpoint inhibition is considered a great success, mostly because it may be applied to many different types of cancer, and differentiation therapy is considered relatively ineffective because it is limited to a few specific cancers. On the other hand, the cost of checkpoint inhibition treatment is 10–20 times more per patient than that of differentiation therapy. Hopefully, future combined treatments and advances in both approaches will increase the effectiveness of these cancer treatments.
Keywords
Introduction
Does cancer arise from dedifferentiation of mature cells, by block of differentiation of immature cells (maturation arrest), or both? 1 This has been a long-standing question that is critical for how cancers might be treated. If cancers arise from mature cells, then therapy or prevention could be directed to how to prevent dedifferentiation. On the other hand, if cancer arises from stem or progenitor cells, then therapy or prevention could be directed to removal of the block in differentiation (differentiation therapy (DT)). 2 Normal tissue renewal involves lineages of cells that mature to terminally differentiated cells so that the number of cells in a normal organ remains essentially the same over time. Cancer arises when there is a block in tissue maturation with accumulation of cells that do not terminally differentiate and continue to proliferate. Pierce et al. 3 stated that cancer is a problem of developmental biology; Potter 4 expressed this as “oncogeny is blocked ontogeny.” DT is directed to removing whatever blocks maturation and allows cancer cells to mature and die. 5 In practice, DT may be directed both to cancer stem cells (CSCs) and to their progeny.
Teratocarcinoma
The proof of concept for forced differentiation of cancer arises from studies on teratocarcinoma. Pierce and Spears 6 pointed out that most of the cells of a teratocarcinoma are fully differentiated. The cancer cells are often present in only a small proportion of the tumor mass as “embryoid bodies,” an observation first made by Rudolf Virchow in mid-19th century. 7 Such observations led others to propose the “embryonal rest” theory of cancer,8–10 including Cohnheim, 11 a student of Virchow. A modern interpretation of the embryonal rest theory is that tissue-determined stem cells are essentially embryonal rests that can not only produce progeny that differentiates into normal mature tissue but also give rise to cancer. Malignant teratocarcinoma stem cells of mice 12 can be converted into normal differentiated tissue cells if transplanted into a normal blastocyst. 13 In fact, normal-appearing offspring from such blastocysts are chimeras of normal blastocyst cells and transplanted teratocarcinoma cells.14,15 Just how the normal blastocyst controls the malignant properties of teratocarcinoma stem cells remains poorly understood. In natural germinal cell tumor growth, malignant cells can give rise to benign cells. Embryonal carcinoma is composed of totipotent germinal cells. Differentiation of these cells may produce cells of all three germ layers, including yolk sac and placenta. DT is based on this principle—finding out how to force undifferentiated CSCs to differentiate into non-cancerous cells. This may be accomplished in vitro with retinoic acid (RA, vitamin A). RA reacts with nuclear receptors (RAR-RXR) leading to upregulation of disabled 2 (Dab2), p21, p27, and β-catenin, which inhibits cyclins and proliferation and activates differentiation signaling (Tcf) while decreasing telomerase β (induces senescence) in embryonal carcinoma cells. 16 For many years, it was known that RA could induce differentiation of cultured teratocarcinoma cells. This finding led to attempts to apply DT in clinical trials. However, a recent phase II clinical trial of 16 patients has failed to show significant clinical antitumor activity in patients with chemotherapy-refractory germinal cell tumors. 17 The primary treatment of teratocarcinoma of ovary or testes is surgery combined with chemotherapy or radiation if surgery is not ablative. For testicular tumors, treatment is usually three cycles of BEP (bleomycin, etoposide, and cisplatin) or four cycles of EP (etoposide and cisplatin), essentially cytotoxic therapy. In contrast to expectations, DT has not yet proved to be effective. 18
Neuroblastoma
Neuroblastoma is a cancer arising from neural crest stem cells and is the most common malignancy diagnosed in the first year of life.
19
It is notable that many neuroblastomas discovered in the first year of life spontaneously regress, but in children over 18 months of age, neuroblastomas with unfavorable pathology or MYC-N amplification require intensive treatment, including surgery, radiotherapy, chemotherapy, as well as immunotherapy and DT. It has been known for years that RA treatment reduces proliferation and induces differentiation of neuroblastoma cells in vitro.
20
Matthay et al.
21
reported a 10% response rate with increased survival of 2 years for high-risk neuroblastoma (Figure 1) and later found 29% response rate with increased survival of 2 years in comparison with transplantation of autologous bone marrow alone.
22
A recent study on the induction of differentiation of glioma cells in culture
23
concludes, The existence of a subgroup of patients harboring RA-responsive glioma cells amenable to differentiation therapy and stratifying such patients with a functional test is easily achievable. This provides the first step to reassess the potential of RA in the context of personalized medicine.

Kaplan–Meier survival curve of high-risk neuroblastoma patients treated with cis-retinoic acid (RA). There is a 10% response rate with a duration of 2 years (delineated by black vertical bars). 21
Myeloid leukemia
Although DT originally focused on stem cells for teratocarcinoma and epithelial cancers, 2 DT of myeloid leukemias has proven to be a much better model (Figure 2). In 1996, Druker et al. 31 identified a small molecular weight compound, identified as SKI571 that competed with the ATP-binding site of the Bcr-Abl tyrosine kinase, blocked the constitutive activation of the leukemic cells, and allowed them to differentiate. This became known as imatinib (Gleevec®) and could be safely and effectively administered to patients. 32 The clinical application of imatinib has increased the 10-year survival of patients with chronic myeloid leukemia (CML) from 20% to up to 91%. In acute promyelocytic leukemia (APL), there is fusion between the promyelocytic leukemia protein and the RA receptor gene, PML/RARα. The PML/RARα fusion product blocks differentiation at the level of the promyelocyte. PML can be treated with all-trans retinoic acid (ATRA), which upregulates ubiquitin activation enzyme-like protein, causing degradation of the gene fusion product and allowing differentiation and apoptosis of the PML cells. This treatment combined with arsenic trioxide has increased 10-year survival of PML patients from 25% to 70% for 48 months (Figure 3). 33

Stages of maturation arrest, chromosomal translocations, and differentiation treatment of myeloid leukemias. In myeloid leukemia, specific gene translocations are directly responsible for blocking differentiation of cells in the myeloid series. For chronic myeloid leukemia, T9:22, the Philadelphia chromosome 24 (Bcr-Abl, break point region-Abelson murine leukemia viral oncogene homolog 1) results in constitutive activation of a tyrosine kinase that keeps myelocytes from differentiation into polymorphonuclear leukocytes. 25 In acute promyeloid leukemia, the T15:17, PML/RARα translocation inhibits maturation at the promyelocyte level, 26 and in acute myeloid leukemia, various translocations that act to stimulate proliferation or inhibit apoptosis block cells at the hematocytoblast stage. 27 Inhibition of STAT3 and FLT3 extends survival in mouse models,27,28 and a clinical trial of inhibition of FLT3 has also proved beneficial.29,30

Kaplan–Meier survival curves for patients with subacute promyeloid leukemia. Treatment with all-trans retinoic acid alone produces a 50% response rate for 48 months when compared to arsenic alone and a 70% response for 86 months when combined with arsenic. 33
In acute myeloid leukemia (AML), there are various translocations that produce fusion products acting on myeloid stem cells (hematocytoblasts) to maintain stemness. For AML, there appears to be two types of mutations: Class I, which increases proliferation (FLT3, KIT), and Class II, which blocks maturation. So far, the major therapeutic approach in treatment of AML is various doses and schedules of delivery of cytotoxic drugs, such as cytarabine and anthracyclines, but there are continued efforts to improve this toxic and relatively ineffective approach. Potential new targets for therapy are directed toward inhibition of interleukin (IL)-3R activation in a subset of patients with mutant FLT3,34,35 blocking STAT3 (epidermal growth factor receptor (EGFR) and IL-6) signaling pathways 36 and targeting MUC-1 to induce differentiation and increase survival. 37 It remains to be seen whether a subset of AML patients may benefit as much as other myeloid leukemia patients. It is quite likely that, as in the case of AML, identification of maturation arrest mutations or translocations in other cancers will be identified and appropriate blocking agents and effective treatments developed, for example, blocking pathways known to maintain stemness, such as Wnt, Oct-4, Notch, BMP, JAK, and others. 38
Comparison of DT and checkpoint inhibition
Antibodies to checkpoint inhibitors block the action of CTLA439,40 and PD-141–43 produced by CD3+ T-cytotoxic cells as off signals to prevent cancer-reactive T cells from proliferating and inhibiting tumor immunity. For example, PD-1 inhibitors have enabled patients with untreatable tumors, such as melanoma and lung cancer, to survive for years, but this effect is not seen in most patients. A recent review of 23 trials of treatments targeted to PD-1 or PDL1 show a response rate from 5% for sarcoma to over 80% in Hodgkin’s disease with a response around 20% for most cancers, including non-small-cell lung cancer and melanoma. 44 In addition to the percent response rate, a critical factor is the duration of response. By multiplying the percent response by the duration, the survival score can be obtained.
Kaplan–Meier curves and survival scores
The analysis of results of clinical trials depends on Kaplan–Meier survival curves. 45 In the study by Herbst et al. 46 shown in Figure 4, the percentage of patients treated with anti-PD-1 alive during the time of study is compared to the percent remaining that is treated conventionally with docetaxel. The top curve (A) shows the results for patients whose cancer cells are greater than 50% PD-1+ by immunofluorescence; the lower curve (B) shows the results for all patients. The most complicated part of this type of analysis is to account for the patients who drop out of the study and are lost to follow-up. 45 Such patients are removed from the study (censured). The small vertical lines on the curves in Figure 4 denote censured patients. The percent response is determined by the number of patients left in the study when the number of patients has become too low to be meaningful. In the study shown, this occurs at 15 months. The duration of the response is when there is a clear separation of the curves. Thus, in Figure 4, the response rate for A is 30% and the duration is 10 months. The survival score is obtained by multiplying the percent response rate by the duration of response. Thus, in A, this is 30 × 10 = 300. For curve B, the response rate is 20% and the duration is 8 months, for a survival score of 160 (20 × 8). A caveat for the survival score is that it gives equal weight to percent patient response and duration of response. This is an arbitrary choice that may not be agreed upon by everyone.
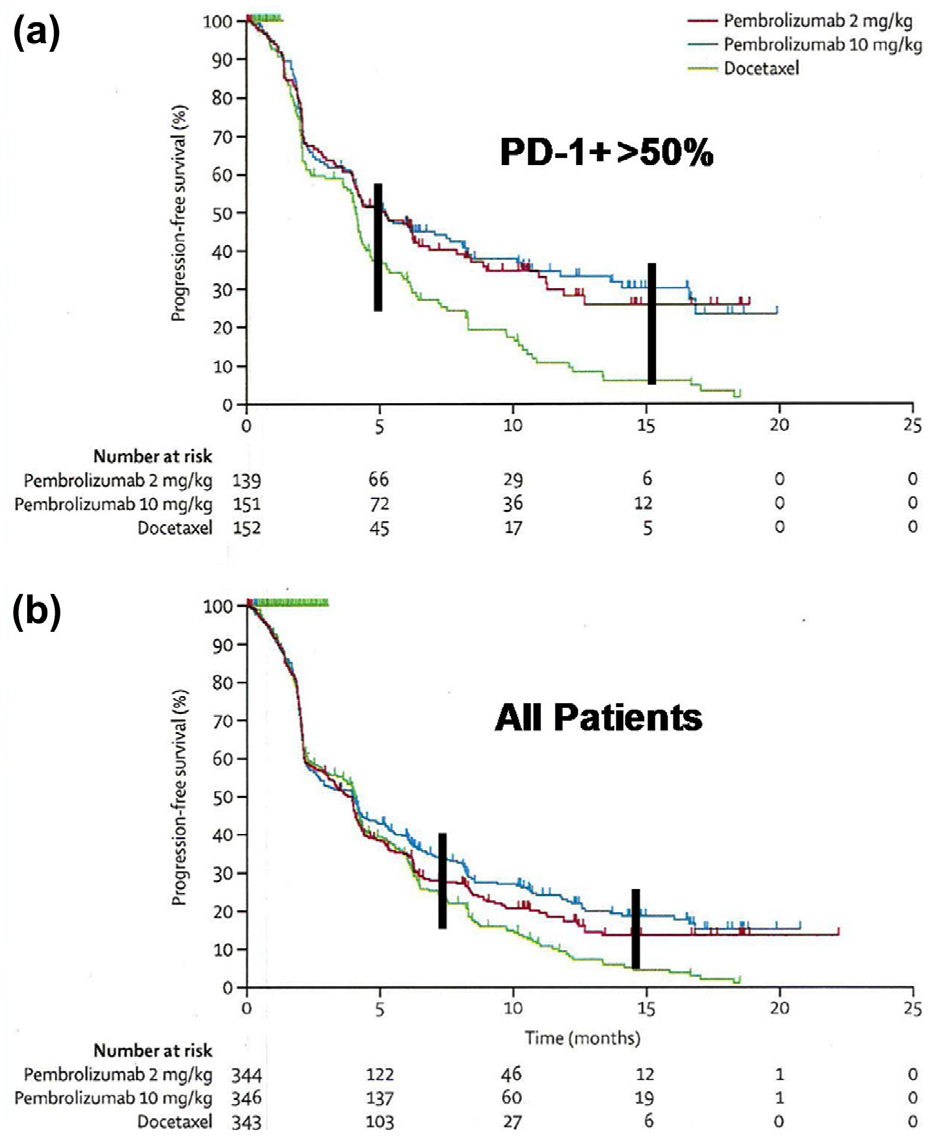
Kaplan–Meier survival curves for anti-PD-1 therapy of non-small-cell lung cancer. (A) Patients with >50% PD-1+ cells and (B) all patients. 46
Comparative survival scores
Some representative survival scores are listed in Table 1. There are three levels for DT: very high for leukemias (2000–6000), moderate for neuroblastoma (240–480), and low for non-small-cell lung carcinoma (NSCLC) (survival score 64). Checkpoint inhibition (CPI) therapy has similar but somewhat lower levels: high for Hodgkin’s lymphoma (1145); 47 moderate for melanoma, 48 renal cell carcinoma,43,49 Merkel-cell carcinoma, 50 and relapsed hematologic cancers (180–512); 51 and variable or low for NSCLC (80–420).52–54 Statistical comparison of the combined treatment response for DT versus CPI was p = 0.0114 by non-paired t-test. The lower than expected scores for CPI treatment of NSCLCs is almost certainly related to the fact that most studies have been done on previously treated patients. This comparison is compromised by the attempt to compare effects of different treatments for different diseases with different natural histories. However, this approach does allow a comparison of overall treatment outcomes.
Representative survival scores for patients treated with DT compared to CPI therapy.
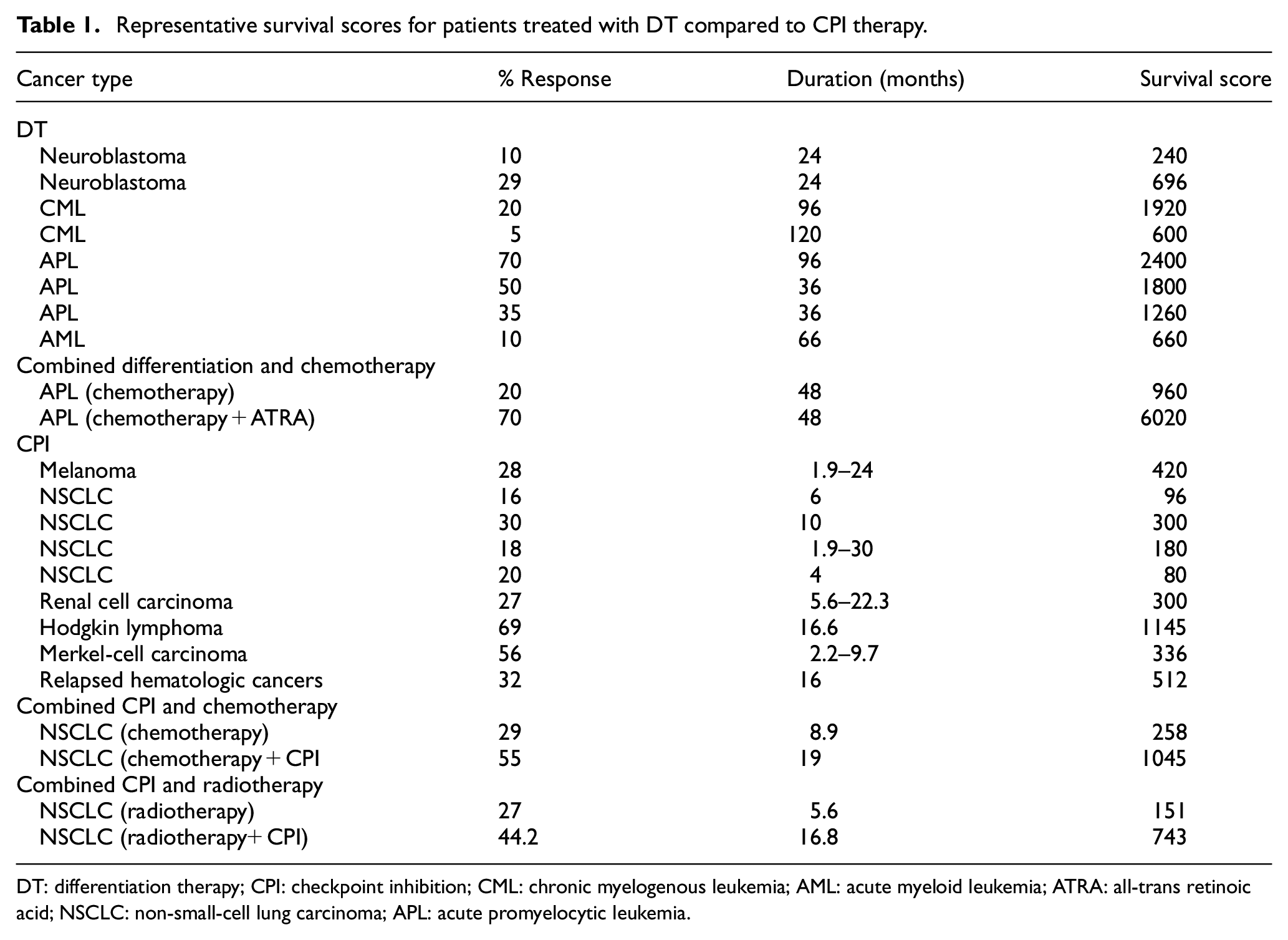
DT: differentiation therapy; CPI: checkpoint inhibition; CML: chronic myelogenous leukemia; AML: acute myeloid leukemia; ATRA: all-trans retinoic acid; NSCLC: non-small-cell lung carcinoma; APL: acute promyelocytic leukemia.
Improving checkpoint inhibitor treatment
Patient selection
Prediction of which patients will respond to checkpoint inhibitors is critical to selection of therapy. 55 First, for patients with cancers who express PD-L1, the receptor for PD-1 will respond to PD-1 blocking therapy better than those who do not, 56 but PD-L1 expression does not always accurately predict which patients will respond to CPI. Second, tumors with CD8+ T-cell infiltrate will respond better than tumors that do not. It is theorized that tumors that have low CD8+ T cells require more aggressive therapy, such as combined anti-PD-1/ anti-CTLA-4 treatments (see below). In addition, since transforming growth factor (TGF)-β inhibits T cells from entering tissues, combined treatment with CPI and the TGF-β blocker, M7824, is being tested. Third, another marker is the tumor mutational burden (TMB). Since each mutation in a tumor may generate a neoantigen, the more mutations that a tumor has, the more likely it is to respond to CPI. Fourth, the gut microbiome may affect CPI outcome. An ongoing study of the effect of an oral treatment with a mix of microbes that match that from anti-PD-1 responders will test this hypothesis.
Other cancer targets (NKG2D and IL-1R8)
The cell surface molecule, natural killer group 2D (NKG2D), is highly expressed on natural killer (NK) cells and T cells. NKG2D reacts with a major histocompatibility complex (MHC)-like class I ligand, which is upregulated in cells under stress. 57 This ligand–receptor interaction is a major pathway for the detection and elimination of damaged, infected, or transformed cells. 58 The NKG2D ligand is highly expressed on cancer cells and virus-infected cells, but not widely on normal tissue cells, 59 so this reaction has potential as a new target for immunotherapy. 57 Expression of NKG2D ligands on the surface of cancer cells correlates with a good prognosis, whereas soluble NKG2D ligand indicates a poor prognosis and negatively impacts anti-PD-1 CPI therapy. Both chemotherapy and radiation increase the expression of NKG2D ligand on cancer cells and enhance tumor immunity. Monoclonal antibodies to NKG2D increase presentation of tumor antigens to activate T cells as well as promote antibody-dependent cellular cytotoxicity of tumor cells by NK cells, neutrophils, and macrophages through Fc receptors. 59 A variety of agents, most prominent being the histone deacetylase (HDAC) inhibitor valproic acid, increase membrane NKG2D expression, whereas others can decrease secretion of non-membrane NKG2D. These have been proposed as potential treatments for cancer.57,60
Interleukin-1 receptor family member R8 (IL-1R8), through binding to IL-37, negatively regulates interleukin receptor and toll-like receptor signaling and inhibits both innate and adaptive immunity. 61 IL-8R binding and activation are active in almost all inflammatory and infectious reactions where it serves to limit inflammation. 61 Colon tumors express less IL-8R than normal colonic mucosa cells and may thus be better able to escape tumor-reactive cells. The possible effect of IL-1R8 in resistance to CPI or as a target for therapy remains to be determined.
Combination therapy
The effect of combination therapy for both DT and CPI is shown in Table 1. Combined ATRA DT and arsenic trioxide improves the survivor score by sixfold over arsenic trioxide alone. There are approximately 2000 clinical trials now underway using six Food and Drug Administration (FDA)-approved humanized monoclonal antibodies to CTLA-4, PD-1, and PL-L162 for cancer treatment for various cancers including melanoma, multiple myeloma, leukemia, lymphoma, glioblastoma, gastric, renal cell, bladder, colorectal, hepatocellular, breast, non-small-cell lung, prostate, and other cancers.62,63 However, as indicated above, about 60%–70% of patients do not respond to single-agent CPI therapy (Table 1). This result has stimulated the use of checkpoint inhibitors in combination with conventional treatments, 64 such as cisplatin, 5-fluorourcil, and lenalidomide, 65 as well as targeted therapy and radiation therapy. 65 Chemotherapy may increase tumor antigenicity, disrupt immune suppression, enhance T-cell responses, or increase antigenicity or major histocompatibility marker expression. FDA approval of combinations of pembrolizumab (anti-PD-1) and chemotherapy has resulted in multiple new clinical trials. 66 The early results of a combination trial of pembrolizumab (anti-PD-1) and carboplatin and pemetrexed for NSCLC patients resulted in 55% response rate for 13 months (survival score 715) compared to 29% response rate for 6 months (survival score 132) for pemetrexed alone 67 with updated survival time of 19 months (survival score 1045) for combination compared to 8.9 months (survival score 258) for pemetrexed. 68 In a phase III trial for squamous NSCLC, combining pembrolizumab with taxene and carboplatin resulted in a survival score of 928 for combined therapy versus 168 for chemotherapy. 69 Similar results have been found in other clinical trials. 66 However, the combined chemo-immunotherapy approach is just beginning and much needs to be done to determine the optimal dose/scheduling program for therapy. Table 1 indicates that the survival score for combined therapy (1045) is much better than chemotherapy alone (258) or CPI alone (80–300).
Molecular targeted therapy (signaling pathways) and CPI therapy
In melanoma, BRAF mutations are associated with decreased antitumor immunity, elevation of PD-L1 and accumulation of regulatory T cells, and downregulation of MHC-1, 69 which can at least temporally be reversed by BRAF inhibitors. The combination of checkpoint inhibitors, ipilimumab (anti-CLTA-4) and nivolumab (anti-PD-1), with kinase (MAPK) inhibitors and inhibitor of BRAF mutation resulted in tumor growth inhibition, 70 but other combined studies have been complicated by severe liver and gastrointestinal toxicities. Much more work is needed to determine whether this approach is beneficial. Vascular endothelial growth factor (VEGF) stimulates tumor vascularization and is immunosuppressive. Multiple clinical trials are currently underway in breast, gynecological, lung, and other cancers, using bevacizumab and pembrolizumab, inhibitors of VEGF. 71 These show some promising findings, such as an increase in intra-tumoral T cells, but definitive positive therapeutic effects have not yet been convincingly demonstrated. 66 An additional target for combination therapy is CD-40.72,73 CD-40 is a co-stimulatory signal that binds to CD-40L and enhances the immune response to tumors. Anti-CD40 is an agonist for CD-40 that activates this pathway and is being tested in combination with nivolumab (anti-PD-1).
Radiotherapy
Radiotherapy induces cell death and can stimulate antitumor immunity. A clinical trial of chemoradiation followed by durvalumab (anti-PD-L1) in stage III NSCLC resulted in a response rate of 44.2% for 16.8 months (survival score 743) compared to 27% for 5.6 months in those receiving chemoradiation and placebo (survival score 151) or anti-PD-1 alone (80–300), an encouraging early result. 74
Adverse effects
From 5% to 87% of patients treated with checkpoint blockade experience grade 3–4 treatment–related adverse effects with up to 100% having less serious effects.44,75 Serious effects include pneumonitis, hepatitis, colitis, endocrinopathy, and nephritis that may require cessation of treatment. In contrast, with RA treatment of APL or neuroblastoma, only mild dryness of the lips and skin with occasional headaches and digestive symptoms have been reported.76,77
Cost
The estimated cost per cancer patient per year for CPI therapy is US$1,000,000, 78 compared to US$150,000 for Gleevec 79 and US$60,000 for RA plus arsenic trioxide for myeloid leukemia. 80
Conclusion
Based on survivor scores, DT is somewhat better than CPI therapy. However, CPI can be applied to many different types of cancer, whereas DT is limited to neuroblastoma and myeloid leukemias. There should be continued search for specific agents for DT, as well as the possibility of combining DT with CPI. The limiting factor is the relatively small number of cancer types that can be successfully treated with DT. On the other hand, CPI treatment is roughly 10–20 times more expensive than DT and has much more severe adverse effects.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.