
Abstract
Colorectal cancer is the second and third most common cancer in men and women, respectively, worldwide. Alterations such as genetic and epigenetic are common in colorectal cancer and are the basis of tumor formation. The exploration of the molecular basis of colorectal cancer can drive a better understanding of the disease as well as guide the prognosis, therapeutics, and disease management. This study is aimed at investigating the genetic mutation profile of five candidate microRNAs (hsa-miR-513b-3p, hsa-miR-500b-3p, hsa-miR-500a-3p, hsa-miR-450b-3p, hsa-miR-193a-5p) targeted by seven genes (APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS)) using in silico approaches. Two datasets (dataset 1 from our previous study and dataset two (The Cancer Genome Atlas, Nature 2012) were considered for this study. Protein–protein interaction, expression analysis, and genetic profiling were carried out using STRING, FireBrowse, and cBioPortal, respectively. Protein–protein interaction network showed that epidermal growth factor receptor has the highest connection among the target genes and this can be considered as the hub gene. Relative to other solid tumors, in colorectal cancer, six of the target genes were downregulated and only CASP8 was upregulated. Genes with protein tyrosine kinases domain were frequently altered in colorectal cancer and the most common alteration in these genes/domain are missense mutation. These results could serve as a lead in the identification of driver genes responsible for colorectal cancer initiation and progression. However, the intense mechanism of these results remains unclear and further experimental validation and molecular approaches are the focal points in the nearest future.
Introduction
One of the fundamental processes driving the initiation and progression of colorectal cancer (CRC) is the accumulation of a variety of genetic and epigenetic changes in epithelial cells of the colorectum. 1 Over the past decade, major advances have been made in the understanding of cancer epigenetics, particularly regarding aberrant DNA methylation, microRNA and noncoding RNA deregulation, and alterations in histone modification states. The number of driver events required for human tumorigenesis has remained one of the fundamental issues in cancer research. 2 Gene mutations have long been known to be important in cancer formation. 3 However, epigenetic alterations have only recently been recognized as significant contributors to cancer development. Genomic sequencing of malignancies from cohort studies will advance researchers’ understanding of the genomic landscape and insight into their cancer pathogenesis and treatment. 4 However, limited information regarding the demographics, epidemiology, and clinical annotations for cancer cases has been a major setback for complex genomic exploration. 5 Another notable drawback to cancer sequencing study is the limited statistical power to significantly identify mutated genes that have an intermediate or lower frequency of mutation (e.g. <5% frequency). Cancer driver genes can be extensively studied by increasing the sample number sequenced in each disease type, mostly in CRC. 6 Genomic exploration and cancer molecular profiling are useful tools to unravel potential tumor biomarkers. 7 The prevailing consensus suggests that epigenetic alterations in CRC occur early and manifest more frequently than genetic alterations. 1 The expression of microRNAs has different effects on the molecular mechanism of carcinogenesis of the gastrointestinal tract from adenoma to carcinoma. The wide range of expression patterns of these microRNAs in CRC found in tumors and healthy tissues could be associated with diagnosis and prognosis. Circulating microRNAs have also been considered as diagnostic and prognostic biomarkers following their correlation with CRC.
From our previous study, 5829 genes were targeted by these microRNAs. 8 These targets were identified using three different databases (miRDB, TargetScan, and mirDIP) according to the microRNA sequences (candidate and validated) and the following criteria: miRNA 3′ site, Conservation Status, and the seed region.9–12 The resulting lists generated were queried separately and further analyzed by the intersection analysis with R-package (https://cran.r-project.org/) to obtain a unique gene list after the removal of redundancies by CD-HIT. Furthermore, DAVID (Database for Annotation, Visualization, and Integrated Discovery) database was employed to identify genes associated with CRC from differentially expressed gene list. The genes were defined as differentially expressed genes that were significantly related to CRC at p < 0.05 (final gene list; p < 0.05). Based on the prioritization and function assignment, the top seven genes, namely, APC, KRAS, TCF7L2, epidermal growth factor receptor (EGFR), insulin-like growth factor receptor (IGF1R), CASP8, and GNAS were considered for the genomic profiling study in this article. Different cells can express hundreds of copies of certain microRNA alone, and cancer cells are not excluded. Efforts have been made to identify CRC-specific microRNA transcripts as prospective diagnostic biomarkers which can be used to develop precancerous stage detection.13,14 In addition, advances in genomic technologies have led to the identification of a variety of specific epigenetic alterations as potential clinical biomarkers for CRC patients. This research briefly investigates the frequency of alterations of the microRNA target genes (APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS) in colorectal adenocarcinoma (TCGA, Nature 2012) patients/samples, and details of the field regarding the distribution of mutation across protein domains, and clinical usefulness of various alterations as biomarkers for the management of CRC patients.
Materials and methods
Data selection
In this study, two datasets were queried using the cBioPortal, with the first dataset being the differentially expressed microRNA target genes from our previous in silico studies.8,15,16 Seven of the genes targeted by our candidate microRNAs that showed significance (Figure 1) were profiled and their expression in CRC was further evaluated. The Colorectal Adenocarcinoma in The Cancer Genome Atlas (TCGA) Colorectal Cancer project (TCGA, Nature 2012) consisting of the whole-exome sequencing in colorectal carcinoma tumor/normal pairs was used as the second dataset. 17

(a) Pictorial representation of dataset 1 and (b) schematic overview of the analyses performed. The orange nodes represent candidate microRNAs; the white nodes represent their specific target genes. Statistically significant microRNA target gene network analysis was built using the Esyn software at http://www.esyn.org/builder.php?type=Graph.
Analysis of microRNA target genes for cancer genomics
cBioPortal is an open-access resource for exploring multidimensional genomics data. The database substantially reduces the drawback between complex genomic data and cancer researchers in order to provide high-quality access to molecular profiles and their clinical attributes. This further provides easy result interpretation of rich datasets into biologic insights and clinical applications. 18 The cBioPortal database was used to query multiple genes (dataset 1) in a single dataset (dataset 2) (Figure 1(b)). The portal was accessed at http://www.cbioportal.org/index.do for Cancer Genomics. The genomic datasets were queried using the interactive web interface with the option to query a single cancer study to explore the relevant genomic profile in the microRNA target genes in CRC samples.
Protein–protein interaction network
STRING (Search Tool for the Retrieval of Interacting Genes) accessed at http://string-db.org/ is a unique tool, equipped to provide a comprehensive view of all the known and predicted interactions and associations among proteins. This database was used to construct a protein–protein interaction (PPI) network of the candidate microRNA target genes.
Expression analysis of the target genes
FireBrowse is a web-based expression analysis tool that provides easy and refined means to investigate the TCGA cancer data, using a powerful computational infrastructure, application programming interface (API), graphical tools, and online reports. This database sits above the TCGA-Genomic DNA Affinity Chromatography Firehose, one of the extensive and most effectively characterized web-based cancer datasets in the world. 19 MicroRNA target gene expression in some solid tumors and in corresponding normal tissues was analyzed using data from TCGA. Data analysis was performed using FireBrowse (http://firebrowse.org/), which provides access to analyze data generated by TCGA. From the interface, colorectal adenocarcinoma was selected as the cohort after which microRNA target genes were submitted individually to generate an expression profile between a tumor and normal tissues among 12 solid tumors (Table 1). RNA-seq by expectation-maximization (RSEM) was selected as the data format with the expression sorted alphabetically.
List of abbreviations used in graphical result from FireBrowse solid tumors.

Statistical analysis
The parameter of interactions in STRING was set as the highest confidence >0.900. Degree of association of the candidate microRNA target genes was considered using both Spearman’s and Pearson’s correlation coefficient and test of goodness of fit (R 2 > 0.250). A p < 0.05 was considered to be statistically significant.
Data availability
The datasets and the clinical data were obtained from the online databases as described above in the “Materials and methods” section. cBioPortal, STRING, and FireBrowse were accessed from the web-server.
Results
Dataset identification
The seven microRNA target genes obtained from our previous study (Figure 1(a)) were used alongside data from TCGA as query in order to explore the genomic profiles of these genes following the workflow presented in Figure 1(b). The criteria for target genes selection was based on gene prioritization, functional assignment, and association with CRC using bioinformatics approaches and the p value was set at 0.05 for significance. The final gene list considered in this study includes APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS.
The genetic landscape of the candidate microRNA target genes
It is crucial to correlate genomic mutations and gene expression patterns in clinical stratification of CRC patients. The cBioPortal cancer genomics tool was used to evaluate the genetic alterations of the microRNA target genes in CRC patient samples. Genetic alterations of APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS (e.g. mRNA expression (upregulation and downregulation) and copy number alterations (deletion and amplification)) were evaluated in the TCGA dataset (TCGA, 2012) to determine their association with colorectal pathogenesis and progression. From the oncoprint, 179 cases (92%) were alteration in at least one of the seven microRNA target genes, with the frequency of alteration in each of the seven genes as shown in Figure 2. It was observed that of the seven candidate genes, APC was the most frequently altered gene (77% with 150 cases altered) followed by KRAS (43% with 83 cases altered), TCF7L2 (18% with 36 cases altered), GNAS (13% with 26 cases altered), IGFIR (8% with 16 cases altered), EGFR (7% with 13 cases altered), and CASP8 (6% with 12 cases altered). The alterations observed include truncating mutations, deep deletion, missense mutation (putative driver), and amplification.

Genetic landscape (Oncoplot) of microRNA target genes in 195 colorectal adenocarcinoma (TCGA, Nature 2012) patients/samples. The figure is a schematic representation (Oncoplot) of the most commonly mutated microRNA target genes. Each column represents a patient. Colors depict the type of mutations for each gene. Complete samples (195 patients/samples). Queried genes are altered in 179 (92%) of the queried patients/samples.
Genetic profiles of microRNA target genes in CRC using cBioPortal
The mRNA expression of the microRNA target genes was plotted against copy number alterations using cBioPortal (Figure 3). Inactivation of the microRNA target genes was observed to be due to mutation, amplification, and deletions. However, these alterations are not the only way to decrease the levels of functional protein in a cell. Colorectal adenocarcinoma (TCGA, Nature 2012) database was selected to observe the relationship between APC (Figure 3(a)), KRAS (Figure 3(b)), TCF7L2 (Figure 3(c)), EGFR (Figure 3(d)), IGF1R (Figure 3(e)), CASP8 (Figure 3(f)), and GNAS (Figure 3(g)) copy number alterations and mRNA levels. Five alterations (deep deletion, shallow deletion, diploid, gain, amplification) were detected in this study. Six out of seven (APC, KRAS, TCF7L2, EGFR, IGF1R, and CASP8) of the microRNA target genes present more gains and less deletions, and were mostly diploid in colorectal carcinoma. GNAS mRNA is increased in the samples in which GNAS is gained and amplified. These results indicated that the mRNA expressions of the target genes (APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS) are associated with genetic status (deletion or amplification) in CRC.
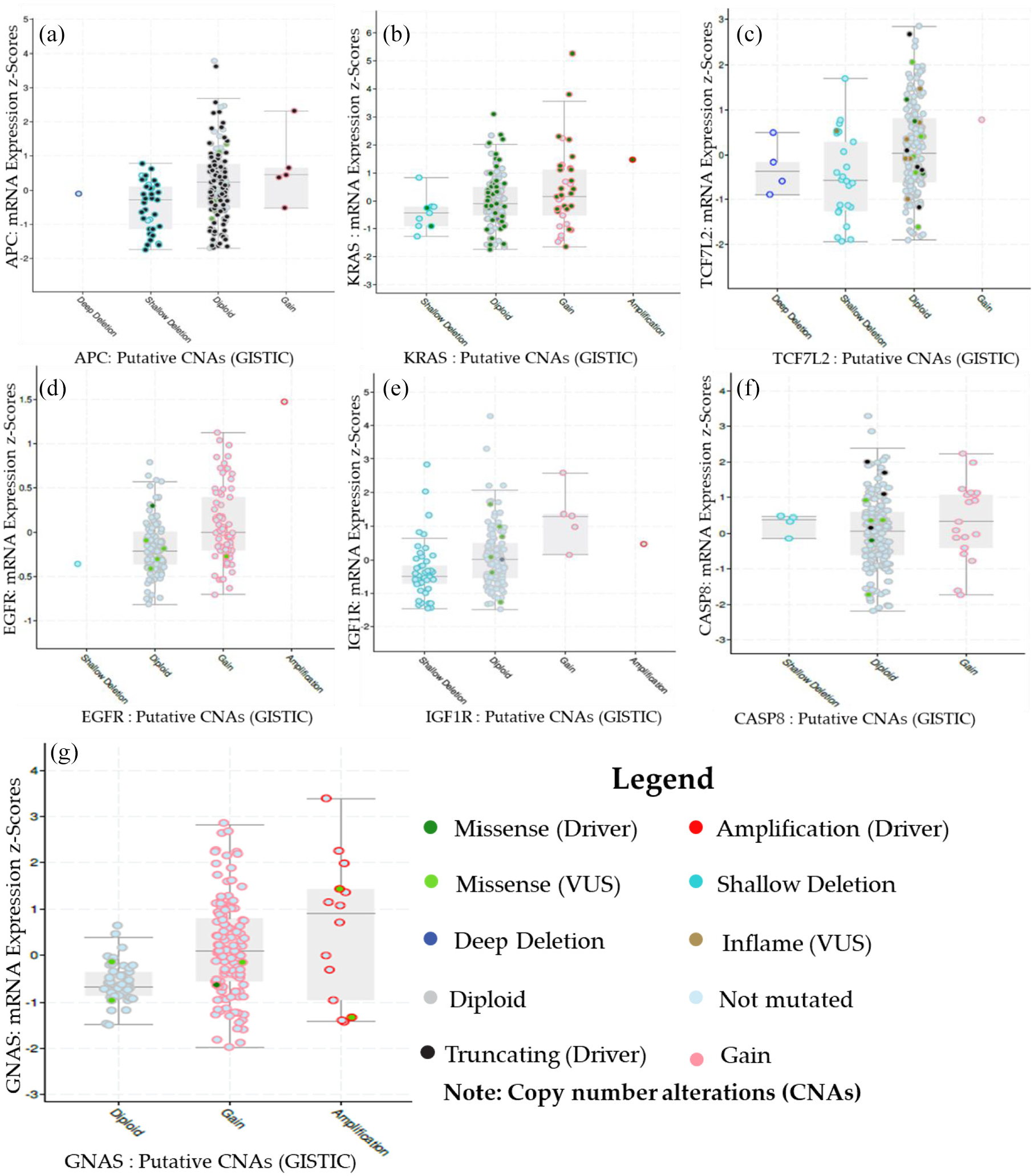
Genetic profiles of microRNA target genes in CRC using cBioPortal. Relative expression levels as a function of relative microRNA target gene copy numbers were plotted against colorectal adenocarcinoma (TCGA, Nature 2012). Shallow deletion – heterozygously deleted; Diploid – two alleles present; Gain – low-level gene amplification event; Amplification – high-level gene amplification event.
Co-expression/correlation analysis
The graphical representation and the statistical results of the co-expression analysis are presented in Figure 4 and Table 2. The graphs analyzed the mRNA expression of each gene in the colorectal adenocarcinoma samples and statistically ranked the degree of association of genes using the test of goodness of fit, Spearman’s rank, and Pearson’s correlation coefficient (Figure 4(a)–(g), Table 2). These assess the linear relationships and monotonic relationships between microRNA target genes and the co-expressed genes. The closer it is to zero, the weaker the association. The result identified seven co-expressed genes that are positively correlated with the target genes in our study (p < 0.05). Functions of the microRNA target genes in CRC may be further explored through APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS.

Co-expression analysis of the query genes. The box plot shows the log2 mRNA expression level between microRNA target genes (X-axis) and co-expressed genes (Y-axis). The solid line indicates linear fit, r, Pearson’s correlation coefficient and p-values indicate significance of correlation. A p-value of < 0.05 was considered statistically significant.
MicroRNA target genes and their correlated genes.

Note: Only positively correlated genes were selected with a p value <0.05 (cBioPortal).
Survival analysis
The association between microRNA target genes and survival in CRC patients was studied using the survival feature in cBioPortal. The figure shows the two curves; cases with alteration(s) in query microRNA target genes and cases without alteration(s) in query genes. The survival curve indicated that CRC patients with microRNA target genes amplification had no significant association between the microRNA target genes and overall survival (p = 0.964, Figure 5) in 2 years. However, it could be seen from the figure that the survival rate was increased when the microRNA target genes were upregulated. Survival analysis is crucial in cancer research as the majority of cancer studies investigate time to event endpoints. This is equally used to define prognostic indices for mortality or recurrence of a disease, and to study the outcome of treatment. The survival plot was used to estimate the survival probability of the candidate microRNA target genes in CRC patients with overall survival as the endpoint. This was done by comparing the survival rate between cases with alteration in query genes and its counterpart at the end of the time curve of the survival analysis. Although the association with survival analysis is not significant, it could be observed that patients with microRNA target genes alteration have a better survival.
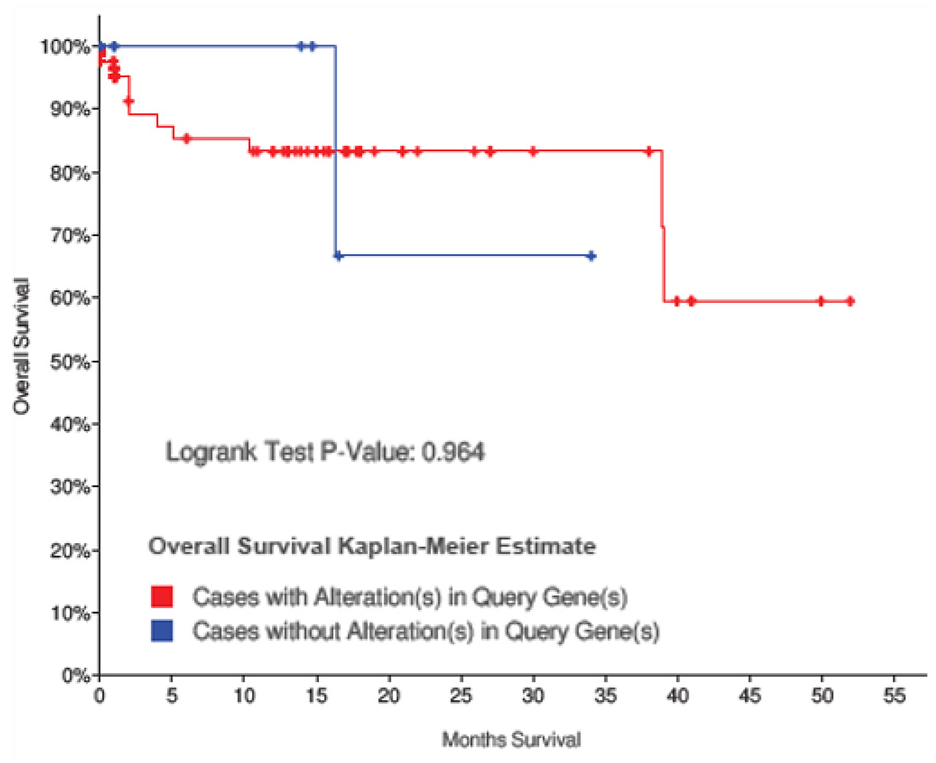
The survival analyses of CRC cases, with and without alterations in microRNA target genes (cBioPortal database).
MicroRNA target genes mutation hotspot analysis
The mutation hotspots of the target genes were analyzed using the cBioPortal web-based tool. Mutations are drawn as a lollipop along with the domain structure of the gene. The height of the lollipop reflects how many times the mutation was detected. Each lollipop shows the hotspot for specific gene mutations in CRC. The graphical summary showed the position and frequency of all the mutations in the context of Pfam protein domains encoded by the canonical gene isoform (Figure 6). The integration of heterogeneous evidence is an important step toward personalized cancer treatment to catalog mutational hotspots that are biologically and therapeutically relevant and thus represent where targeted therapy would likely be beneficial. 20 However, existing methods do not sufficiently portray varying functionality of individual mutations within the same genes. The mutations hotspot provided details as a graphical illustration of all nonsynonymous mutations identified in each query gene.

Distribution of mutations of microRNA target genes across protein domains in CRC.
PPI network
In order to establish the biological roles of the microRNA target genes, the STRING web-based tool was used to establish a PPI (Figure 7). The network consists of 7 nodes and 12 edges with an average node degree of 3.43 and the average local clustering coefficient of 0.767 at PPI enrichment p value of 5.25 × 10–05. EGFR was considered as the hub gene connecting five nodes followed by KRAS and IGF1R, which were connected by four nodes. Other genes were connected by three nodes except TCF7L2 which interacted with only APC. The majority of the interactions are either experimentally validated or curated from the database.

Protein–protein interaction (PPI) network of the significant microRNA target genes. Nodes represent the genes while edges represent PPI. The color of the edges indicates varying PPI evidence.
This information, therefore, suggests novel directions for further research and provides cross-species predictions for effective interaction mapping.
Gene expression analysis
To understand the mechanism underlying the dysregulation of the microRNA target genes in CRC, their expression levels were investigated using FireBrowse web-based tool (Figure 8(a)–(g)). Data mining showed that APC, KRAS, TCF7L2, and EGFR were significantly downregulated in COREAD than in normal colorectal tissue as opposed to the expression of CASP8 and GNAS, but the expression of IGF1R is relatively the same in both COREAD and normal tissue (Figure 8). Relative to other solid tumors, in CRC, the expressions of six of the microRNA target genes, namely, APC, KRAS, TCF7L2, EGFR, IGF1R, and GNAS, are relatively low. But GNAS expression in COREAD was significantly increased in CRC compared with other solid tumors.

Differential plots of microRNA target genes in selected solid tumors in TCGA. Expressions of (a) APC, (b) KRAS, (c) TCF7L2, (d) EGFR, (e) IGF1R, (f) CASP8, and (g) GNAS. Sample number (N) = 626 for all tumor samples and 51 for all the normal tissues. Red bars indicate tumor, blue bars indicate normal, and white bars indicate missing samples.
Discussion
Colorectal carcinogenesis is a multistep process that results from the accumulation of numerous genetic alterations. The activation of multiple signaling pathways plays an important role in regulating cell proliferation, angiogenesis, cell motility, and apoptosis.21,22 One of the major goals of cancer genomics is the identification of the driver genes responsible for tumor initiation and progression. The microRNAs targeted genes in this study were carried using the in silico approach to detect the frequency of mutation in each gene and investigate the frequency of mutation distribution across all protein domains present in the nucleotide sequences of the genes. The microRNA target genes were used as dataset due to the significance of the associated microRNAs from our previous study (hsa-miR-513b-3p, hsa-miR-500b-3p, hsa-miR-500a-3p, hsa-miR-450b-3p, hsa-miR-193a-5p) along their validated microRNAs hsa-miR-193a-5p, hsa-miR-450b-3p, hsa-miR-501-3p, hsa-miR-501-3p, and hsa-miR-513a-3p). 8 After the assignment of function and the determination of the mechanism of action to these microRNAs and target genes (previous studies),15,16,23 their genomic profiling deserves closer attention in CRC.
The integration of heterogeneous evidence is an important step toward personalized cancer treatment to catalog mutational hotspots that are biologically and therapeutically relevant and thus represent where targeted therapy would likely be beneficial. 20 However, existing methods do not sufficiently portray varying functionality of individual mutations within the same genes. The mutations hotspot provided details as a graphical illustration of all nonsynonymous mutations identified in each query gene. The graphical summary showed the position and frequency of all mutations in the context of Pfam protein domains encoded by the canonical gene isoform.
The mRNA expressions of the target genes are associated with genetic status (deletion or amplification) of APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS in CRC. The cross-reference between the microRNA expression profiles and the expression profiles of its target genes can provide an effective viewpoint to understand the regulatory functions of the microRNA. Functions of the microRNA target genes in CRC may be further explored alongside the associated genes. Survival analysis is crucial in cancer research as the majority of cancer studies investigate time-to-event endpoints. This is equally used to define prognostic indices for mortality or recurrence of a disease, and to study the outcome of treatment. The survival plot was used to estimate the survival probability of the candidate microRNA target genes in CRC patients with overall survival as the endpoint. This was done by comparing the survival rate between cases with alteration in query genes and its counterpart at the end of the time curve of the survival analysis. Although the association with survival analysis is not significant, it could be observed that patients with microRNA target genes alteration have a better overall survival.
Although CRC is regarded as one of the most common malignant diseases worldwide, 24 its involvement in signaling pathways and driven-genes is not completely elucidated. 25 The integration of heterogeneous evidence is an important step toward personalized cancer treatment to catalog mutational hotspots that are biologically and therapeutically relevant and thus represent where targeted therapy would likely be beneficial. 20 However, existing methods do not sufficiently portray varying functionality of individual mutations within the same genes. The mutations hotspot provided details as a graphical illustration of all nonsynonymous mutations identified in each query gene. From the mutation point of view, APC is a tumor suppressor gene involved in the Wnt signaling pathway. 26 There are 30 missenses and 90 truncation mutations in the sample (not shown). For the missense mutation, R1450 is commonly altered although the APC_basic domain appears to be frequently mutated. The APC R1450 is recurrently altered in CRC and it is likely to be oncogenic with loss of function as its biological effect with a somatic mutation frequency of 76.4% (percentage of sample with a somatic mutation in APC). KRAS, a GTPase that functions as an upstream regulator of the MAPK and PI3K pathways, is frequently mutated in a diverse range of cancers including colorectal cancers. G12D is the most common missense altered with the somatic mutation frequency of 41.0% and the Ras: Ras family (5–164) appeared to be frequently mutated. This mutated amino acid was identified as a recurrent hotspot (p < 0.05) and a 3D clustered hotspot in a population-scale cohort of tumor samples of various cancer types.27,28 The KRAS G12D mutation may be oncogenic with loss of function. The prognostic implications for KRAS mutations vary between cancer types but have been shown to be associated with poor outcome in CRC, non-small cell lung cancer, and others. 29 Also, the frequency of alteration observed in the novel microRNA gene target is supported by Roa et al. 30 who reported similar frequency of alteration in KRAS in CRC samples. TCF7L2 is regarded as a tumor suppressor and transcription factor. This gene is altered by mutation or deletion in various cancer types, most frequently in CRC. CTNNB1_binding and HMG-H domains are the most frequently mutated with K483_K485del and C486vfs*8 as the most common missense mutation (in-frame deletion) with other mutations such as in-frame deletion, non-stop, fusion, and frameshift deletion. The oncogenic function of K483_K485del mutation is considered unknown. This mutated amino acid was identified as a recurrent hotspot (statistically significant) in a population-scale cohort of tumor samples of various cancer types. 27 EGFR plays a vital role in promoting cell growth. 31 The protein tyrosine kinase is the most frequently mutated domain with L861Q as the most common alteration. The major type of mutation found in this gene is missense. The EGFR L861Q mutation is known to be oncogenic and the biological effect is the loss of function. 32 The existing literature reported that overexpression of EGFR is estimated to be 60%–80% of the tumor and is associated with poor prognosis in CRC. 33 IGF1R is involved in numerous regulatory networks to control developmental and physical functions. 34 The protein tyrosine kinase domain is most frequent. The most common type of mutation observed in this gene is missense. IGF1R encodes the type I insulin-like growth factor receptor which activates downstream pathways involved in growth and cell survival. Therapeutic strategies aimed at targeting the insulin-like growth factor signaling system has demonstrated limited success thus far. 35 Overexpression of IGF1R has been linked to CRC metastasis. Data suggested that IGF1R is an attractive therapeutic target for several tumor types including CRC.36,37 Shiratsuchi et al. 38 also suggested that the expression of IGF1R may be linked to increasing tumor size in CRC. Other research also confirms the association of this gene with CRC.39,40 CASP8 plays a crucial role in the apoptotic pathway and its aberrant expression may lead to cancer. This gene also plays an important role in the defense mechanism against hyperproliferation and tumorigenesis. 41 CASP8 mutation showed that the domain commonly mutated is peptidase_C14 (caspase domain) with the frequency of alteration found in R432*. CASP8, a tumor suppressor, and pro-apoptotic protein are inactivated by mutation or deletion in various cancer types. The CASP8 R432* mutation is likely oncogenic and its biological effect is the loss of function. Qiliu et al. 42 through the meta-analysis study suggested that CASP8 could be a candidate gene for CRC susceptibility. Also, in the late stage of CRC, mutation in this gene may lead to loss of apoptotic function contributing to pathogenesis. 43 The only mutation observed in this gene is missense along with the G-alpha domain. Mutations of this gene have been shown to activate the adenylate cyclase gene and lead to constitutive cAMP signaling. 44 Zauber et al. 44 also reported that GNAS mutations may function as an important driver mutation during certain phases of colorectal carcinogenesis, but may then be lost once the biological advantage gained by the mutated gene is no longer necessary to sustain or advance tumor development.
Based on expression on the target genes, the inactivation of APC is required as an early event to develop most adenomas and carcinomas in the colon and rectum. 45 This gene has been investigated as the most frequently mutated and known driver gene in CRC. 46 Several authors have also reported the expression analysis of this gene to be downregulated in CRC. Birnbaum et al. 26 confirmed that APC status may not be used in the prediction of metastasis and for staging. Schell et al. 46 demonstrated the prognostic role of APC and suggested that this gene may be a good potential therapeutic assignment for CRC. The RAS oncogene has a well-defined role in cell growth and regulation, and its protein product affects many cellular functions including cell proliferation, apoptosis, migration, fate specification, and differentiation. 47 KRAS has been reported to be the most frequently altered gene, with mutations occurring in 17%–25% of all cancers, while its mutations in CRC lead to resistance to select treatment strategies. 48 From this study, the expression KRAS was downregulated in CRC. This gene encodes for a guanosine triphosphate/guanosine diphosphate-binding protein that is downstream of EGFR in the RAS/RAF/MAPK pathway. 49 Studies have also shown that approximately 40% of patients with CRC carry a KRAS mutation. 17 Therefore, APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS are important for the diagnosis of cancer subtype. TCF7L2 is an essential element of the Wnt signaling pathway, which acts as a transcriptional factor in the nucleus.50,51 Its significance and protein expression in the progression of various malignancies is yet to be exploited. 52 The expression analysis of IGF1R did not show any significant difference between normal colorectal tissue and the cancer subtype. IGF1R has been investigated to be a target by miR-448 in CRC and also, its expression was upregulated in CRC tissues and cell lines, 53 which counter the result obtained by this study. Studies have suggested that mutation in GNAS may play a direct role in mucin production.54,55 The EGFR is a transmembrane glycoprotein and receptor tyrosine kinase that is encoded by the c-erbB-1proto-oncogene. 56 This gene is overexpressed in many types of cancers, specifically CRC. 33 EGFR is estimated to be overexpressed in 60%–80% of tumors and is associated with a poor prognosis. 57 The expression analysis of this gene showed that it was downregulated in CRC. Spano et al. 58 revealed that EGFR remains a controversial prognostic factor, whose expression may play an important role in a decision to initiate treatment. Another study also confirmed that the expression of this gene is implicated in CRC pathogenesis. 59 Therefore, it could be a potential target for CRC treatment. Intestinal epithelium that possesses one of the highest renewal rates among human tissues, expresses IGF1R in addition to other genes, and the levels of these receptors are higher in CRC relative to colonic mucosa.60–62 Limited reports have been recovered from this gene to date in CRC. CASP-8 is one of the crucial factors (intrinsic signaling pathway) employed in apoptosis. 63 The expression of this gene (CASP-8) has been reported to be associated with poor prognosis in stages II and III of CRC. 64 Therefore, their functional polymorphism may alter cancer risk. Although several studies have reported that CASP8 may not contribute to the risk of CRC,65–67 this gene deserves a closer look. This information, therefore, suggests novel directions for further research and provide cross-species predictions for effective interaction mapping.
Conclusion
The analysis of the seven microRNA target genes in CRC from our previous study and data from TCGA with combined bioinformatics tools revealed that APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS were frequently altered in CRC. Furthermore, the expression of these genes was correlated with the genomic status. Genes with protein tyrosine kinase domains were frequently altered in CRC and the most common alteration observed in the domain of APC, KRAS, TCF7L2, EGFR, IGF1R, CASP8, and GNAS is missense mutation. The information provided by the expression, overall survival of the patients with alteration in the expression of the mRNA of the target genes, and their genetic mutation hotspots across protein domains in CRC could contribute to a better understanding of the molecular basis of colorectal carcinogenesis, and ultimately propel the transformation of genomic knowledge into clinical practice. These could be further exploited to potentially serve as a resource for explicitly selecting targets for diagnosis and management of CRC. However, the intense mechanism of these results remains unclear, and further experimental validation and molecular approaches are the focal points in the near future.
Footnotes
Acknowledgements
The authors acknowledge the Plant Omics Laboratory and the Bioinformatics Research Group of the University of the Western Cape for their usual support.
Authors’ contributions
All the authors have made significant contributions to the submission of the article. A.F. conceived the concept and the design of the manuscript, and A.K. and A.P. provided the necessary supports and software required for analysis. The analysis and data interpretations were done by both A.F. and A.P., while A.K. and O.O.B. drafted the rough draft and substantively revised the manuscript. Finally, all authors (A.F., O.O.B., A.K., and A.P.) read and approved the submitted version of the manuscript for publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.