
Abstract
MicroRNAs (miRNAs) are small regulatory RNAs that play key gene-regulatory roles in diverse biological processes, particularly in cancer development. Therefore, inferring miRNA targets is an essential step to fully understanding the functional properties of miRNA actions in regulating tumorigenesis. Bayesian linear regression modeling has been proposed for identifying the interactions between miRNAs and mRNAs on the basis of the integrated sequence information and matched miRNA and mRNA expression data; however, this approach does not use the full spectrum of available features of putative miRNA targets. In this study, we integrated four important sequence and structural features of miRNA targeting with paired miRNA and mRNA expression data to improve miRNA-target prediction in a Bayesian framework. We have applied this approach to a gene-expression study of liver cancer patients and examined the posterior probability of each miRNA-mRNA interaction being functional in the development of liver cancer. Our method achieved better performance, in terms of the number of true targets identified, than did other methods.
Introduction
MicroRNAs (miRNAs) are highly conserved small RNAs that have diverse functions, including regulation of cellular differentiation, proliferation, and apoptosis.1,2 These RNA molecules exert their function by inhibiting translation or inducing degradation of their target messenger RNAs. A given miRNA is able to pair with hundreds of transcripts by its seed miRNA nucleotides, allowing it to regulate complex gene-expression programs and induce global physiological changes.
3
Dysfunction of these miRNA molecules has been linked to several human diseases, including different types of cancer. Virtually, all examined tumor types have globally abnormal miRNA expression patterns, where miRNAs play regulatory roles as potential oncogenes or oncosuppressor genes.4–6 Genome-wide profiling showed that about half of miRNA genes are localized in cancer-related genomic regions or fragile loci,
7
where mutations, deletions, or amplifications occur in many human tumors. These observations indicate that miRNAs are candidate genes for tumorigenesis and cancer progression. An essential step and major challenge to understanding the functions of miRNAs in cancer is identification of their target genes. Many computational and experimental approaches have been used to improve the reliability of miRNA-target prediction. TargetScan,8–10 PicTar,
11
DIANA-microT,
12
miRanda,
13
and TargetS14,15 are examples of computational approaches that are based on an analysis of miRNA and mRNA sequences. Generally, they use the following principles to predict miRNA targets:
Seed matches: the Waston-Crick pairing between the 5’ region of the miRNA centered on nucleotides 2–7 and the 3’ untranslated region (UTR) of the target mRNA. Degree of conservation: a functional miRNA target is preferentially conserved across multiple species. Thermodynamic stability, measured by the hybridization energy between miRNA and its candidate target site. It is believed that the total free energy of a functional targeting must be thermodynamically favorable, ie, negative valued. Accessibility energy, which is the free energy required to unpair the nucleotides on the target site to make the target accessible to the miRNA. Target site context, including local AU content; the target position within 3’ UTR; and the residue pairing at 3’ of the putative target site.
9
These computational methods, which integrate multiple types of sequence and structural features, however, have low specificity and a high number of false positives for miRNA-target prediction. More importantly, predictions based on sequence and structural features only represent static miRNA-mRNA interactions. It is not clear to what extent these predicted interactions align with functional miRNA regulation in a particular phenotype or pathological condition. Thus, expression profiling has been proposed as an important information resource for discovering miRNA targets under different conditions. On the basis of this idea, some novel approaches have been developed to predict miRNA targets by integrating expression data into sequence-based prediction. Among them are GeneMiR++,16,17 TaLasso, 18 HOCTAR, 19 BLasso, 20 MAGIA, 21 and HCTarget. 22 They mainly use paired miRNA and mRNA expression data from the same set of samples to refine the sequence-based prediction results and obtain more reliable miRNA targets. However, these approaches do not consider the full spectrum of available sequence and structural features of putative miRNA targets. Instead, they view all potential targets in sequence-based prediction results as equally biologically meaningful. Recently, Xu et al systematically evaluated the effects of sequence and structural features on miRNA-target prediction using the pSILAC dataset as a benchmark.14,15 It was found that all these features were important for improving the accuracy of miRNA-target identification.
In this study, we combined the paired expression data of miRNAs and mRNAs from liver cancer patients, and the sequence and structural features of miRNA targeting to improve miRNA-target prediction. Our approach was based on a Bayesian linear regression model coupled with the Markov chain Monte Carlo (MCMC) algorithm. It uses both sequence and structural feature information to establish a prior probability of a miRNA-target interaction being functional, and paired miRNA and mRNA expression data to compute the likelihood of a putative miRNA-target interaction. By combining these two sources of information, our Bayesian method allows us to effectively sample from the large search space of putative miRNA-mRNA interactions and compute the posterior probability of each putative miRNA target. It represents a powerful means of reconstructing miRNA-mRNA interaction networks, specifically in liver cancer samples, and might help us uncover the mechanisms of tumorigenesis and progression in liver cancer.
Methods
Given a set of expression data of miRNA and mRNA, we modeled the interaction between miRNAs and target mRNAs using a linear model. The log-conditional likelihood function of data can be written in the following form, assuming a normal distribution:
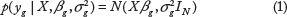
Without additional sequence and structural feature information
The goal of this analysis is to identify a small subset of miRNA–mRNA interactions that are biologically meaningful. In the framework of variable selection,23,24 an indicator matrix is defined as
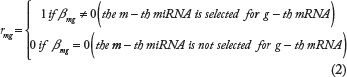
Here, 
Here π can be regarded as the proportion of the true targets in databases.
We used a non-informative prior for β

The joint posterior distribution is written as

To efficiently search the parameter space of 
where 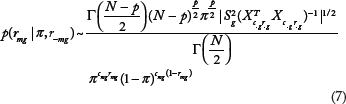
Because rmg is binary, we can define its marginal distribution as a Bernoulli distribution with a success probability of

Here, we implemented a Gibbs sampler to sample each
With Additional Sequence and Structural Features as Prior Information
To incorporate the sequence and structural features of miRNA-target sites into the model, we introduced an 
We further specified a hyperprior on
Following the Gibbs sampling of 
where
Results
We studied the regulatory roles of miRNAs in a dataset of matched miRNA and mRNA expression data for 125 patients with liver cancer from The Cancer Genome Atlas (TCGA). We log-transformed the expression data to ensure that they approximately followed normal distribution during the data preprocessing step. The computationally predicted miRNA–mRNA interactions were extracted from TargetScanHuman (release 6.1) 10 and mapped to the expression dataset. 10 This yielded 67,798 interactions between 170 human miRNAs and 4973 mRNA transcripts, which were used as the prior information for our first model without the addition of miRNA features.
To determine the effects of the additional sequence and structural features of miRNA on target prediction, we obtained context scores and aggregated probability of conserved targeting scores for each miRNA–mRNA pair from TargetScanHuman. The probability of conserved targeting score is a target site conservation score and has been used to measure the degree of miRNA target sequence conservation across multiple species. We also calculated the thermodynamic stability (Δ
In our Bayesian framework without additional miRNA features, the parameter π of the Bernoulli distribution reflects the prior belief about the proportion of true targets in the computationally predicted miRNA–mRNA interactions. Since there were 67,798 putative miRNA–mRNA interactions included in the liver cancer dataset, we set π = 0.07, indicating that 7% of putative interactions are true; thus, the expected number of miRNA–mRNA interactions for each mRNA would be approximately equal to 1. In our model with the miRNA sequence and structural features, we tuned the prior probability of each interaction according to the values of their corresponding features. We set the hyperparameters for the gamma distribution of weights as

Trace plots for (A) the number of selected miRNA-mRNA interactions and (B) the log-posterior probability along the number of iterations.
To assess the performance of our approach, we evaluated the enrichment scores of the results from experimentally validated miRNA–mRNA interactions. If the top-ranked miRNA and mRNA interactions identified from an algorithm include more experimentally validated targets, this algorithm will be considered to have better performance because more predicted interactions can be validated. Here, we extracted the experimentally validated target information from TarBase 6.0, which includes more than 65,000 manually curated, experimentally validated miRNA-gene interactions from eight species.
26
To examine the overlaps between the TarBase information and our prediction results, we mapped all miRNAs in our dataset to the miRNA families in TarBase using the annotations in miRBase. In the liver cancer expression dataset, 609 miRNA–mRNA interactions have been biologically verified. We found that 68 and 79 of these interactions, without and with the addition of miRNA features, respectively, overlapped with the top 5000 targets detected by our model; the well-known GenMiR++ method only identified 56 interactions. On the basis of this observation, we obtained the numbers of false positives and false negatives, and calculated the corresponding statistical significance of the number of true targets identified by different methods using the hypergeometric distribution. For a given number of identified true targets, the smaller the
Enrichment values of experimentally validated targets obtained from our feature-dependent model (feature-MCMC), the model without additional features (non-feature), and the GenMiR++ method. In the top 5000 or 500 predicted interactions, the numbers of experimentally validated targets (true positives), false positives, false negatives, and enrichment significance (

Predicted experimentally validated targets obtained from our feature-dependent model (feature-MCMC), the model without additional features (non-feature), and the GenMiR++ method.

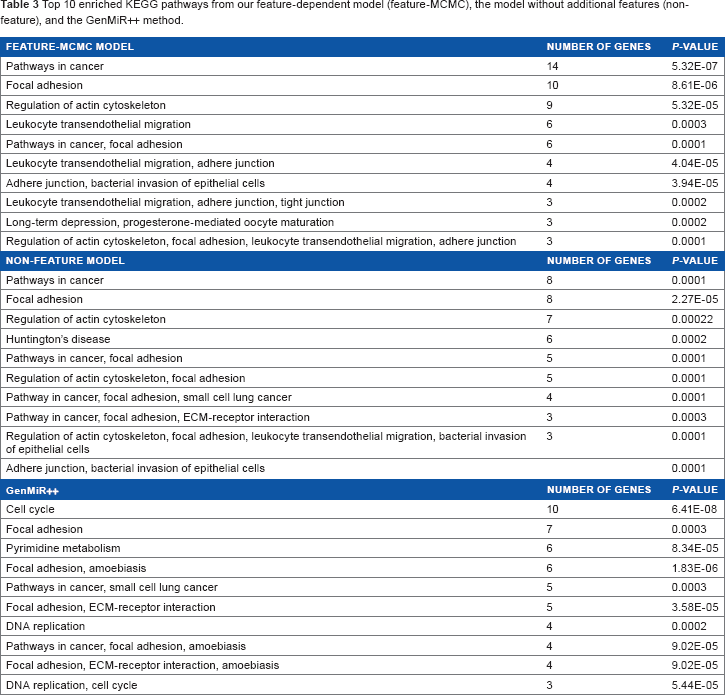
To further investigate the function of our predicted targets and the potential regulatory roles of miRNA in patients with liver cancer, we analyzed the biological relevance of the target genes in a KEGG pathway enrichment study. 27 We used the KEGG pathway annotation to measure the enrichment of the top 200 genes predicted by different methods using the GeneCodis 3.0 tool. As a result, several KEGG pathways were found to be significantly enriched in the results obtained from our models and GenMiR++ (Table 3). Both of our models (with and without additional features) resulted in significantly enriched pathways related to cancer and focal adhesion. For GeneMiR++, the two most prominent pathways were related to cell cycle and focal adhesion.
Top 10 enriched KEGG pathways from our feature-dependent model (feature-MCMC), the model without additional features (non-feature), and the GenMiR++ method.
Among the miRNAs shown in Table 2, hsa-miR-145 and hsa-miR-21 are key regulators during hepatocellular carcinoma genesis.28,29 In particular, hsa-miR-145 functions as a tumor suppressor in liver cancer by targeting the chromatin modification enzyme, histone deacetylase.
28
In this study, we discovered many novel functional targets of hsa-miR-145, including MUC1. We used the pair hsa-miR-145-MUC1 to illustrate the effectiveness of our model. We grouped liver cancer patients by their hsa-miR-145 expression level (higher or lower than average). The patients in the high hsa-miR-145 group had significantly lower MUC1 expression than those in the other group (the

Down-regulatory effect of hsa-miR-154 on MUC1.The liver cancer patients were grouped according to their hsa-miR-145 expression levels (higher or lower than average). The cumulative distribution of the MUC1 expression levels in these two groups of patients was plotted, respectively (high hsa-miR-154, red dashed line; low hsa-miR-154, blue solid line). The
Conclusions
In this study, we integrated matched miRNA and mRNA expression data with the sequence and structural features of miRNA seeds to improve miRNA target prediction. Compared to previous approaches, 25 our model restricts our search space to the putative miRNA targets obtained from a well-known miRNA target prediction database; thus, our model led to significantly less computational complexity but higher target prediction specificity. In addition, using a Bayesian linear regression model, we successfully incorporated four key features of miRNA-mRNA interactions; each assigned a different weight by the MCMC sampling procedure, as the prior knowledge in our model. Our investigation of paired miRNA and mRNA expression profiles in liver cancer patients successfully demonstrated the advantages of our feature-dependent model. Our results showed that the top interactions identified by our feature-dependent model are significantly more enriched in experimentally validated targets and are more biologically meaningful than are those identified by the GenMiR++ method or the model without additional feature information.
In addition, with the recent intensive research in this field, a large body of experimentally verified miRNA target information has accumulated in available databases, such as StarBase 30 and miRWalk. 31 There is a strong interest in leveraging this information to improve target prediction sensitivity and accuracy, and this will be the focus of our future work. From a Bayesian perspective, we expect to be able to easily incorporate this information by assigning different prior distributions to the information sources according to their reliability, as in a proposed prior Lasso framework. 32
Author Contributions
Conceived and designed the experiments: ZW, YL. Analyzed the data: ZW, WX, HZ. Wrote the first draft of the manuscript: ZW, YL. Contributed to the writing of the manuscript: ZW, YL. Agree with manuscript results and conclusions: ZW, XW, HZ, YL. Jointly developed the structure and arguments for the paper: ZW, YL. Made critical revisions and approved final version: ZW, XW, HZ, YL. All authors reviewed and approved of the final manuscript.