
Abstract
Chemotherapy-induced neuropathy is a highly problematic, dose-limiting effect of potentially curative regimens of cancer chemotherapy. When neuropathic pain is severe, patients often either switch to less-effective chemotherapy agents or choose to discontinue chemotherapy entirely. Conventional chemotherapy drugs used to treat lung and breast cancer, multiple myeloma, and lymphoma include paclitaxel, vincristine, and bortezomib. Approximately 68% of patients receiving these anticancer drugs develop neuropathy within the first month of treatment, and while strategies to prevent chemotherapy-induced neuropathy have been investigated, none have yet been proven as effective. Recent reports suggest that chemotherapy-induced neuropathy is associated with signal transduction molecules, including protein kinase C and mitogen-activated protein kinases. It is currently unclear whether protein kinase C inhibition can prevent chemotherapy-induced neuropathy. In this study, we found that tamoxifen, a protein kinase C inhibitor, suppressed paclitaxel-, vincristine-, and bortezomib-induced cold and mechanical allodynia in mice. In addition, chemotherapy drugs induce neuropathy via the protein kinase C/extracellular signal-regulated kinase pathway in the spinal cord in lumbar segments 4–6 and dorsal root ganglions. In addition, tamoxifen was shown to act synergistically with paclitaxel to inhibit tumor-growth in mice injected with tumor cells. Our results indicated that paclitaxel-, vincristine-, and bortezomib-induced neuropathies were associated with the protein kinase C/extracellular signal-regulated kinase pathway in the lumbar spinal cord and dorsal root ganglions, which suggest that protein kinase C inhibitors may be therapeutically effective for the prevention of chemotherapy-induced neuropathy when administered with standard chemotherapy agents.
Keywords
Introduction
Chemotherapy-induced peripheral neuropathy is a common and debilitating side effect of anticancer drugs such as oxaliplatin, paclitaxel, vincristine, or bortezomib. 1 Neuropathy resulting from chemotherapy can be painful and disabling, causing a significant loss of functional abilities and decreasing the quality of life. Neuropathy is the predominant reason for dose modification and discontinuation of treatment, and may thereby affect overall survival. 2 Paclitaxel-induced neuropathy is positively correlated with increasing number of doses per cycle, total cumulative dose, and duration of infusion; grade 3 or 4 sensory neuropathy occurs in 20%–35% of patients receiving 250 mg/m2 paclitaxel every 3 weeks. 3 Vincristine-induced neuropathy is cumulative and dose-dependent, and initially occurs at doses of 4–10 mg. 4 Bortezomib-induced grade 1 or 2 neuropathy occurs in 25%–33% of patients with newly diagnosed multiple myeloma and in 27%–75% of patients with recurrent multiple myeloma.5,6 Treatments for neuropathy using drugs such as gabapentin, lamotrigine, or pyridoxine plus pyridostigmine have not shown efficacy in random clinical trials.7,8 As chemotherapy becomes more effective, the number of cancer survivors is likely to increase and neuropathy will become an increasingly important factor affecting the cancer survivors’ quality of life. Thus, it is of critical importance to elucidate the mechanisms underlying paclitaxel-, vincristine-, and bortezomib-induced neuropathy and to develop effective treatment strategies for this debilitating syndrome.
Transient receptor potential (TRP)-family proteins play critical roles in pain sensation. This family consists of six members, namely, TRP canonical (TRPC), TRP vanilloid (TRPV), TRP ankyrin (TRPA), TRP melastatin (TRPM), TRP polycystin (TRPP), and TRP mucolipin (TRPML).8 TRPA1 and TRPV1 were reported to be involved in paclitaxel-induced neuropathy, while TRPA1 antagonists have been shown to suppress bortezomib-induced mechanical and cold hypersensitivity.9,10 In addition, enhanced thermal and mechanical sensitivity has resulted from the sensitization of TRPA1 and TRPV1 induced by protein kinase C (PKC).11,12 Our previous study indicated that activation of the PKC/extracellular-regulated protein kinase (ERK) pathway contributed to oxaliplatin-induced neuropathy. 13 Furthermore, ERK1/2 signaling downstream to the toll-like receptor 4 in a rat dorsal root ganglion (DRG) was involved in paclitaxel-induced peripheral neuropathy. 14 It was also reported that activation of mitogen-activated protein kinases (MAPKs), including ERK1/2, p38MAPK, and c-Jun N-terminal kinase, in the spinal cord contributed to vincristine-induced neuropathy. 15 These findings suggest that neuropathy associated with chemotherapy may be mediated by PKC/ERK pathway activation in the spinal cord.
Tamoxifen, a selective estrogen receptor modulator and PKC inhibitor, is widely used in estrogen-positive breast cancer therapy. Previous studies have reported that tamoxifen suppresses PKC activation and phosphorylation of its downstream signaling molecules in estrogen-negative and estrogen-independent cancer cell lines and that it suppresses oxaliplatin-induced neuropathy.13,16,17 However, the potential efficacy of tamoxifen in suppressing chemotherapy-induced neuropathy other than that induced by oxaliplatin remains unclear. Therefore, we investigated whether administration of tamoxifen suppresses paclitaxel-, vincristine-, and bortezomib-induced neuropathy in a mouse model.
Materials and methods
Mice
All animal studies were approved by the Kindai University Animal Care and Use Committee and performed in accordance with the Kindai University recommendations for the handling of laboratory animals. The ethical procedures followed met the requirements of the United Kingdom Coordinating Committee for Cancer Research (UKCCCR) guidelines (2010).
Male 5-week-old BALB/c mice were purchased from Shimizu Laboratory Animals (Kyoto, Japan). The mice were housed in standard cages that were maintained at 25°C with controlled lighting on a 12 h light/12 h dark cycle, and were allowed free access to water and food pellets.
Drugs
Paclitaxel and vincristine were purchased from Wako (Osaka, Japan). Bortezomib and chelerythrine chloride were purchased from LC Laboratories (Woburn, MA, USA). Tamoxifen was purchased from Sigma (St. Louis, MO, USA). Paclitaxel was dissolved in cremophor/ethanol/saline (1:1:18, Sigma, St. Louis, MO, USA). Vincristine, bortezomib, tamoxifen, and chelerythrine chloride were dissolved in saline containing 0.5% dimethyl sulfoxide (DMSO).
Paclitaxel-, vincristine-, and bortezomib-induced neuropathy models
Mice were treated with 6 mg/kg paclitaxel, 0.2 mg/kg vincristine, 1 mg/kg bortezomib, or saline (vehicle) on days 0 and 7 via intraperitoneal (i.p) injection. Tamoxifen (30 mg/kg) was orally administered 12 h after paclitaxel, vincristine, or bortezomib on day 0 and daily thereafter. Chelerythrine chloride (2.5 mg/kg) was i.p. injected 12 h after paclitaxel, vincristine, or bortezomib on day 0 and daily thereafter. Behavioral tests were performed on days 0, 2, 4, 6, 8, 10, 12, and 14.
Behavioral assays
Behavioral assays were performed as described in a previous study. 13 Briefly, paclitaxel-, vincristine-, or bortezomib-induced cold allodynia and hyperalgesia were assessed using a hot/cold-plate analgesimeter (Ugo Basile, Milan, Italy). In this procedure, mice were acclimated to the testing environment for 1 h, after which they were placed individually in the center of a cold plate maintained at 10°C (cold allodynia) or 4°C (cold hyperalgesia). The time to the first pain-related behavior (jumping or lifting and licking of the hind paw) was recorded, up to a cutoff time of 30 s.
Paclitaxel-, vincristine-, and bortezomib-induced mechanical sensitivity analyses were performed using von Frey filaments (Ugo Basile) to produce forces of 0.16, 0.4, and 1.4 g. Mice were individually placed in 20 × 20 cm2 clear plastic boxes set with metal mesh floors and allowed to acclimatize to the environment for 15 min. For each filament, five stimuli were applied to each hind paw with an interval of 3–5 s between stimuli. The paw withdrawal thresholds of the trials were then averaged. Mechanical sensitivities were scored as follows: 0, no response; 1, paw withdrawal; or 2, immediate flinching of the stimulated paw.
Western blotting
Protein extracts from mouse lumbar spinal cords and DRGs, and subsequent western blotting assays, were prepared as previously described. 13 Extract supernatants were measured using a bicinchoninic acid (BCA) assay kit (Thermo Scientific, Rockford, IL, USA). Protein extracts were separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) into 20 µg samples and then transferred onto polyvinylidene (PVDF) membranes (GE Healthcare, Buckinghamshire, UK). The membranes were blocked using tris-buffered saline containing 3% skimmed milk and incubated overnight at 4°C with each of the following antibodies: phospho-PKCα (ser638), phospho-PKCδ (ser643), anti-phospho-ERK (Thr202/Tyr204), and anti-ERK1/2 (Cell Signaling Technology, Beverly, MA, USA), phospho-PKCε (Thr729; Santa Cruz Biotechnologies, CA, USA), and anti-β-actin antibody (Sigma). Subsequently, the membranes were incubated with horseradish peroxidase-coupled anti-rabbit IgG sheep antibodies (GE Healthcare) for 1 h at room temperature. The reactive proteins were visualized using a Luminata Forte HRP substrate (Merck Millipore, Nottingham, UK) according to the manufacturer’s instructions.
Subcutaneous tumor growth study
In order to generate a breast carcinoma allograft model, 4T1 cells (mouse triple negative breast cancer cell lines) were grown to 80% confluence and trypsinized. Suspensions consisting of single cells with >90% viability were injected subcutaneously (s.c.) into the left flank of each mouse as a bolus of 1 × 106 cells. Treatment with tamoxifen or paclitaxel was started when the tumor reached a size of approximately 50 mm3. Tumors were measured every 2 days with a square caliper and their volumes were calculated using the formula (a × b2)/2, where a and b are the larger and smaller diameters, respectively.
Statistics
All results are expressed as means and S.E.M. of several independent experiments. Statistical comparisons were performed by analysis of variance (ANOVA) with Dunnett’s test for multiple comparisons. Values of p < 5% were considered significant.
Results
Tamoxifen suppressed paclitaxel-, vincristine-, and bortezomib-induced cold allodynia
In order to investigate the protective effect of tamoxifen against paclitaxel-, vincristine-, and bortezomib-induced cold sensitivity, mice were treated with 30 mg/kg tamoxifen daily in addition to receiving paclitaxel, vincristine, or bortezomib. Although paclitaxel, vincristine, or bortezomib alone induced a significant reduction in withdrawal thresholds at 10°C, suggesting the presence of cold allodynia, combined administration of these chemotherapies with tamoxifen significantly improved the withdrawal thresholds, suggesting a reduction in cold allodynia (Figure 1). However, treatment with paclitaxel, vincristine, or bortezomib did not affect withdrawal thresholds at 4°C (cold hyperalgesia) with or without tamoxifen treatment (Figure 2). Mice that received paclitaxel, vincristine, bortezomib, and tamoxifen did not exhibit any weight loss (Supplementary Figure 1).
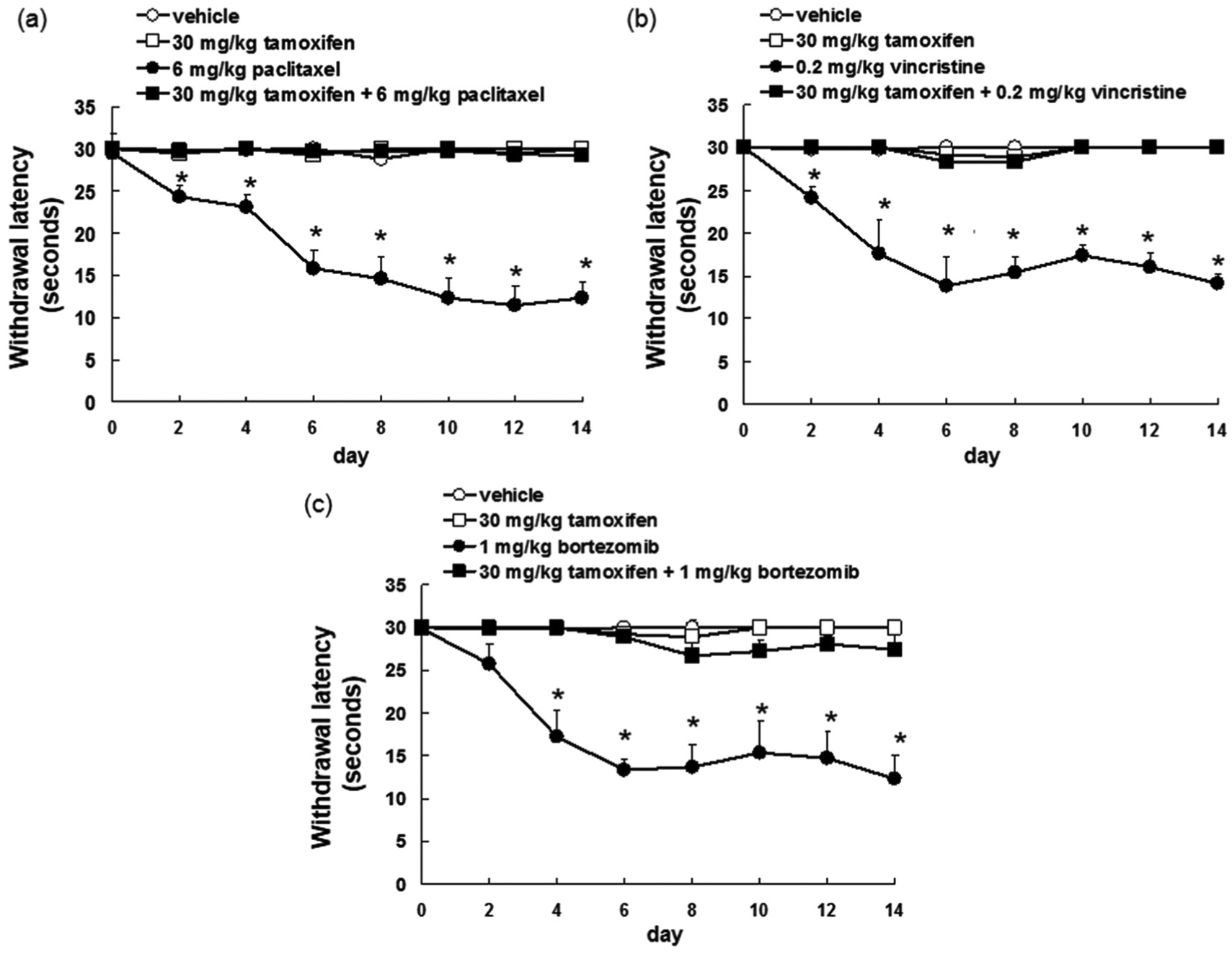
Tamoxifen inhibited paclitaxel-, vincristine-, and bortezomib-induced cold allodynia. (a) Paclitaxel (6 mg/kg, n = 10), (b) vincristine (0.2 mg/kg, n = 15), or (c) bortezomib (1 mg/kg, n = 15) were administrated i.p. weekly for 2 weeks (days 0 and 7). Tamoxifen (30 mg/kg, n = 10–15) was administrated p.o. daily for 14 days. Withdrawal latencies are the mean ± S.E.M. time it took mice to withdraw their hind paws following a cold stimulation (10°C).

Paclitaxel-, vincristine-, and bortezomib did not induce cold hyperalgesia. (a) Paclitaxel (6 mg/kg, n = 10), (b) vincristine (0.2 mg/kg, n = 15), or (c) bortezomib (1 mg/kg, n = 15) were administrated i.p. weekly for 2 weeks (days 0 and 7). Tamoxifen (30 mg/kg, n = 10–15) was administrated p.o. daily for 14 days. Withdrawal latencies are the mean ± S.E.M. time it took mice to withdraw their hind paws following a cold stimulation (4°C).
Tamoxifen suppressed paclitaxel-, vincristine-, and bortezomib-induced mechanical hypersensitivity
Next, we investigated the suppressive effect of tamoxifen on paclitaxel-, vincristine-, and bortezomib-induced mechanical hypersensitivity. This was assessed using von Frey filaments as follows: 0.16 g (mechanical allodynia), 0.4 g (intermediate), or 1.4 g (mechanical hyperalgesia). Tamoxifen suppressed paclitaxel-, vincristine-, and bortezomib-induced mechanical allodynia and hyperalgesia (Figure 3). These observations suggest that tamoxifen suppressed paclitaxel, vincristine, or bortezomib induced neuropathy.

Tamoxifen inhibited paclitaxel-, vincristine-, and bortezomib-induced mechanical allodynia. (a–c) Paclitaxel (6 mg/kg, n = 10), (d–f) vincristine (0.2 mg/kg, n = 15), or (g–i) bortezomib (1 mg/kg, n = 15) were administrated i.p. weekly for 2 weeks (days 0 and 7). Tamoxifen (30 mg/kg, n = 10–15) was administrated p.o. daily for 14 days. (a–i) Number of paw lifts out of five mechanical stimulations using von Frey filaments corresponding to (a, d, g) innocuous (0.16 g), (b, e, h) intermediate (0.4 g), and (c, f, i) noxious (1.4 g) bending forces. The pain threshold is obtained for two lifts (dotted line).
Tamoxifen inhibited PKC/ERK activation induced by treatment with paclitaxel, vincristine, and bortezomib and enhanced the antitumor effect of paclitaxel
Our previous results indicated that tamoxifen suppressed oxaliplatin-induced activation of the PKC/ERK signaling pathway in spinal cords. 13 In order to investigate the inhibitory effect of tamoxifen on paclitaxel-, vincristine-, or bortezomib-induced neuropathy, PKCα, PKCδ, PKCε, and ERK1/2 levels in spinal cord lumber segments 4–6 and DRGs were examined via western blotting. We found that treatment with paclitaxel and vincristine enhanced the activation of PKCα, PKCδ, PKCε, and ERK1/2 in the spinal cord and DRGs (Figure 4(a)–(d)), and that treatment with bortezomib increased PKCα, PKCε, and ERK1/2 activation in the spinal cord and enhanced PKCα, PKCδ, PKCε, and ERK1/2 phosphorylation in DRGs (Figure 4(a)–(d)). In addition, combined treatment of tamoxifen and paclitaxel, vincristine, or bortezomib significantly suppressed phosphorylated protein expression compared to treatment with paclitaxel, vincristine, or bortezomib alone (Figure 4(a)–(d)).

Tamoxifen inhibited the paclitaxel-, vincristine-, and bortezomib-induced activation of PKC/ERK pathway. (a) phosphorylated PKCα (phospho-PKCα), phosphorylated PKCδ (phospho-PKCδ), phosphorylated PKCε (phospho-PKCε), and phosphorylated ERK1/2 (phospho-ERK1/2) protein content analyzed by western blotting in the spinal cord (L4–L6) obtained from mice on day 14 after treatment with paclitaxel, vincristine, bortezomib, or tamoxifen. Loading of equivalent amounts of protein was verified by the relative expression of β-actin. (b) Quantification of the amount of phospho-PKCα, phospho-PKCδ, phospho-PKCε, or phospho-ERK1/2, normalized to the amount of total β-actin or ERK, respectively. The results are representative of four independent experiments. *p < 0.01 versus vehicles. (c) phosphorylated PKCα (phospho-PKCα), phosphorylated PKCδ (phospho-PKCδ), phosphorylated PKCε (phospho-PKCε), and phosphorylated ERK1/2 (phospho-ERK1/2) protein content analyzed by western blotting in the dorsal root ganglion obtained from mice on day 14 after treatment with paclitaxel, vincristine, bortezomib, or tamoxifen. Loading of equivalent amounts of protein was verified by the relative expression of β-actin. (d) Quantification of the amount of phospho-PKCα, phospho-PKCδ, phospho-PKCε, or phospho-ERK1/2, normalized to the amount of total β-actin or ERK, respectively. The results are representative of four independent experiments.
To confirm PKC activation, we administered chelerythrine, a pan-PKC inhibitor, to paclitaxel-, vincristine-, or bortezomib-treated mice to determine if PKC suppression inhibited paclitaxel-, vincristine-, or bortezomib-induced neuropathy. Treatment with chelerythrine (2.5 mg/kg, i.p., daily) suppressed paclitaxel-, vincristine-, or bortezomib-induced neuropathy (Figure 5).

Chelerythrine inhibited paclitaxel-, vincristine-, and bortezomib-induced neuropathy. (a) Paclitaxel (6 mg/kg, n = 6), (b) vincristine (0.2 mg/kg, n = 6), or (c) bortezomib (1 mg/kg, n = 6) were administrated i.p. weekly for 2 weeks (days 0 and 7). Chelerythrine (2.5 mg/kg, n = 6) was administrated i.p. daily for 14 days. Withdrawal latencies are the mean ± S.E.M. time it took mice to withdraw their hind paws following a cold stimulation (10°C). *p < 0.01 versus vehicles (ANOVA with Dunnet’s test). (d–f) Paclitaxel (6 mg/kg, n = 6), (g–i) vincristine (0.2 mg/kg, n = 6), or (j–l) bortezomib (1 mg/kg, n = 6) were administrated i.p. weekly for 2 weeks (days 0 and 7). Chelerythrine (2.5 mg/kg, n = 6) was administrated i.p. daily for 14 days. (d–l) Number of paw lifts out of five mechanical stimulations using von Frey filaments corresponding to (d, g, j) innocuous (0.16 g), (e, h, k) intermediate (0.4 g), and (f, i, l) noxious (1.4 g) bending forces. The pain threshold is obtained for two lifts (dotted line). *p < 0.01 versus vehicles (ANOVA with Dunnet’s test).
We further investigated whether tamoxifen enhanced the efficacy of paclitaxel in a mouse model of breast cancer and found that mice treated with tamoxifen and paclitaxel combination had lower tumor volumes than those treated with paclitaxel alone (Figure 6). This result suggests that tamoxifen enhanced the antitumor effect of paclitaxel. These results indicate that the inhibitory effects of tamoxifen on paclitaxel-, vincristine-, or bortezomib-induced neuropathy occur via the suppression of the PKC/ERK pathway and that tamoxifen enhances the tumor growth-suppressant effect of paclitaxel.
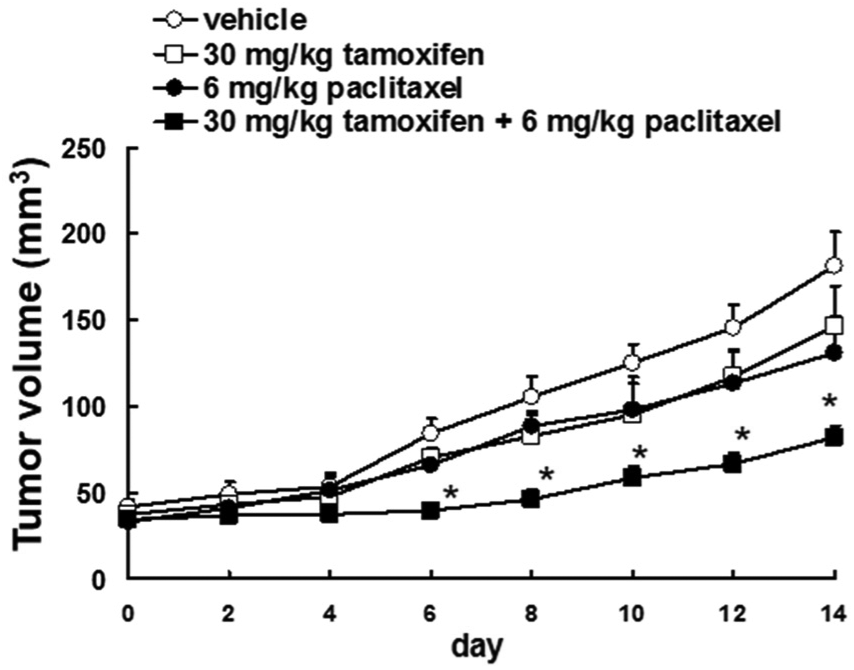
Tamoxifen enhanced the efficacy of paclitaxel on suppressing tumor growth. Paclitaxel (6 mg/kg, n = 10) was administrated i.p. weekly for 2 weeks (days 0 and 7). Tamoxifen (30 mg/kg, n = 10) was administrated p.o. daily for 14 days. Tumor growth over time during administration of paclitaxel or tamoxifen. Tumor volume is presented as the mean ± S.E.M.
Discussion
In this study, we demonstrated that tamoxifen suppresses paclitaxel-, vincristine-, and bortezomib-induced neuropathy via inhibition of the PKC/ERK pathway, and that combined tamoxifen and paclitaxel treatment significantly suppresses the volume of 4T1-based tumors over treatment with paclitaxel alone. Paclitaxel- and bortezomib-induced neuropathies were shown to involve activation of PKCβII, PKCδ, and PKCε in the DRG.18,19 Activation of PKC increased the expression of phosphorylated ERK1/2 in the spinal cord, which has been associated with chronic pain.13,20 It has been reported that sensitization of TRPV1 and TRPA1 through activation of PKCε is involved in paclitaxel-induced neuropathy (cold and mechanical allodynia) in mice 9 and that, mustard oil, a TRPA1 stimulator, induced the transmission of nociceptive signals and the activation of PKC signaling. 12 Here, we provided evidence for the phosphorylation of PKCα, PKCδ, and PKCε in the spinal cord and DRG during vincristine-induced neuropathy. These results suggest that paclitaxel-, vincristine-, and bortezomib-induced neuropathy could be associated with TRP channel activation via PKC signaling. We have previously reported that tamoxifen inhibits PKCα and PKCδ activation in melanoma cells and synergistically potentiates the tumor suppressant effect of oxaliplatin in colon cancers.13,17 Tamoxifen has been shown to induce growth inhibition via the suppression of the mammalian target of rapamycin (mTOR) and the activation of AMP-activated protein kinase in human triple negative breast cancer cells. 21 It was also demonstrated that 4-hydroxytamoxifen, an active metabolite of tamoxifen, suppresses cell proliferation via modulation of Bcl-2 family proteins, downregulation of c-Myc, and upregulation of p27kip1 in multiple myeloma cells. 22 In addition, anti-estrogen therapies such as tamoxifen reduced the sensitivity to paclitaxel via the suppression of pre-mRNA splicing factor 4 kinase expression in estrogen-positive breast cancer cell lines. 23 It was also indicated that tamoxifen and paclitaxel or docetaxel had an antagonistic effect in estrogen-positive breast cancer cell lines, and concurrent treatment with 4-hydroxytamoxifen and paclitaxel yielded less than additive antitumor effects in estrogen-positive breast cancer cells.24,25 These results indicated that tamoxifen may enhance the sensitivity to paclitaxel in estrogen-negative and/or estrogen-independent cancer cells. Taken together, these findings suggest that decreasing PKC/ERK pathway activation could play an important role in both the inhibition of paclitaxel-, vincristine-, and bortezomib-induced neuropathy and in the suppression of tumor growth.
In this study, we found that co-treatment with 30 mg/kg tamoxifen suppressed paclitaxel-, vincristine-, and bortezomib-induced neuropathy. In breast cancer patients, oral administration of 20 mg/kg tamoxifen resulted in an average steady-state plasma concentration of 89.9 ng/mL. 26 In mice, 20 mg/kg daily oral administration of tamoxifen resulted in a mean plasma concentration of 25.9 ± 17.9 ng/mL. 27 These findings suggest that steady-state plasma concentration of tamoxifen in mice is similar to minimum concentrations in humans
In conclusion, our results showed that the activation of the PKC/ERK pathway in the spinal cord and DRG due to treatment with paclitaxel, vincristine, and bortezomib is correlated with chemotherapy-induced neuropathy and that tamoxifen suppressed chemotherapy-induced neuropathy via the inhibition of this pathway. In addition, tamoxifen enhanced the antitumor effect of paclitaxel in a breast cancer-bearing mouse model. These findings indicate that tamoxifen may be a potential suppressant of chemotherapy-induced neuropathy and can be used in combination pharmacological therapy.
Supplemental Material
Supplementary_Figure_legends_20180804 – Supplemental material for Tamoxifen suppresses paclitaxel-, vincristine-, and bortezomib-induced neuropathy via inhibition of the protein kinase C/extracellular signal-regulated kinase pathway
Supplemental material, Supplementary_Figure_legends_20180804 for Tamoxifen suppresses paclitaxel-, vincristine-, and bortezomib-induced neuropathy via inhibition of the protein kinase C/extracellular signal-regulated kinase pathway by Masanobu Tsubaki, Tomoya Takeda, Mikihiro Matsumoto, Natsuki Kato, Shota Yasuhara, Yu-ichi Koumoto, Motohiro Imano, Takao Satou and Shozo Nishida in Tumor Biology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
All animal studies were approved by the Kindai University Animal Care and Use Committee.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by a Grant-in-Aid for Scientific Research (C) (Grant number 15K08116), Grant-in-Aid for Young Scientists (B) (Grant number 16K18965) from the Japan Society for the Promotion of Science (JSPS) and by Ministry of Education, Culture, Sports, Science and Technology (MEXT)-Supported Program for the Strategic Research Foundation at Private Universities, 2014–2018 (Grant number S1411037).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.