
Abstract
Let-7a, miR-34a, and miR-199 a/b have gained a great attention as master regulators for cellular processes. In particular, these three micro-RNAs act as potential onco-suppressors for hepatocellular carcinoma. Bioinformatics can reveal the functionality of these micro-RNAs through target prediction and functional annotation analysis. In the current study, in silico analysis using innovative servers (miRror Suite, DAVID, miRGator V3.0, GeneTrail) has demonstrated the combinatorial and the individual target genes of these micro-RNAs and further explored their roles in hepatocellular carcinoma progression. There were 87 common target messenger RNAs (p ≤ 0.05) that were predicted to be regulated by the three micro-RNAs using miRror 2.0 target prediction tool. In addition, the functional enrichment analysis of these targets that was performed by DAVID functional annotation and REACTOME tools revealed two major immune-related pathways, eight hepatocellular carcinoma hallmarks–linked pathways, and two pathways that mediate interconnected processes between immune system and hepatocellular carcinoma hallmarks. Moreover, protein–protein interaction network for the predicted common targets was obtained by using STRING database. The individual analysis of target genes and pathways for the three micro-RNAs of interest using miRGator V3.0 and GeneTrail servers revealed some novel predicted target oncogenes such as SOX4, which we validated experimentally, in addition to some regulated pathways of immune system and hepatocarcinogenesis such as insulin signaling pathway and adipocytokine signaling pathway. In general, our results demonstrate that let-7a, miR-34a, and miR-199 a/b have novel interactions in different immune system pathways and major hepatocellular carcinoma hallmarks. Thus, our findings shed more light on the roles of these miRNAs as cancer silencers.
Keywords
Introduction
It has been reported that hepatocellular carcinoma (HCC) is caused by various genetic and epigenetic changes that induce multiple oncogenic pathways;1,2 however, the major contributing mechanisms underlying these pathways are still elusive.
Micro-RNAs (miRNAs) are class of non-coding RNAs that act as master regulators of gene expression. They control several cellular pathways through modulating the expression of tens to hundreds of genes which are involved in those pathways. 3 They function by controlling on/off expression switch to fine-tune protein levels. The miRNA action is mediated either by inducing target genes degradation (messenger RNA (mRNA)) or repressing their translation at the post-transcriptional level. 4 These two different modes of action depend on whether the binding sequence of the miRNA is fully or partially complementary to the mRNA-binding site. 5
Liver tissue is enriched with innate and adaptive immune cells that act against inflammatory responses, tumor transformation, and progression.6,7 Examples of these cells include natural killer (NK) and T-cells, which are correlated positively with apoptosis and negatively with proliferation. 8 In addition, liver expresses specific miRNAs, which naturally regulate its immunological processes.9–11 For instance, these miRNAs have been shown to regulate the activation of T- and B-lymphocytes; however, they also work as functional actors in HCC initiation and progression.12,13
The present study is a workflow based on our previous research on the top 26 differentially expressed miRNAs in HCC and their relation to cancer hallmarks.14,15 We focused on three highly represented miRNAs—let-7a, miR-34a, and miR-199a/b—where we collectively predicted their significantly regulated target genes for further understanding of their function in HCC pathways and immune system. These three HCC-associated miRNAs were specifically selected as key candidates for the in silico analysis because of their expression and functionality mounting evidences as detailed below. Let-7a, miR-34a, and miR-199a/b have been known as the most prominent tumor suppressor miRNAs which are evolutionarily conserved and they control basic programs during cellular proliferation and differentiation.3,16–19 However, the deregulation of these miRNAs has been shown to contribute to the development and progression of multiple cancers including HCC due to the gain of function of multiple proto-oncogenes as their miRNA-mediated repression is inhibited.3,20,21
Let-7a has been shown as the fifth most abundantly expressed miRNA in normal liver with an average ratio in miRNome 3.3%. 21 Nevertheless, this miRNA was found to be significantly downregulated in a group of HCC tissues and cell lines by more than 50% as compared to adjacent normal liver.21–24 MiR-34a has been shown to be significantly downexpressed or silenced in 76% of HCC tissues as compared to adjacent normal tissues.3,25–30 Besides, miR-34a has been determined among the miRNA signatures, which is remarkably associated with HCC invasion and metastasis.25,31 MiR-199a/b has been shown as the third most highly expressed miRNA in normal liver with an average ratio in miRNome 4.9%. 21 However, these two miRNAs have been found to consistently decrease in HCC tissues, as much as 82.5% and 73%, respectively, and this decrement is significantly correlated with poor survival of patients.32–36
Let-7a expression restoration in HCC tissue has been found to inhibit tumor formation and growth; this is due to targeting multiple components of cell proliferation pathways. For instance, introduction of let-7a precursor into HepG2 cells could significantly decrease cell proliferation by 30% and increase apoptosis.20,23,37,38 Moreover, let-7a has been evolved to post-transcriptionally control innate and adaptive immunity (e.g. production of interleukin 13 (IL-13)).39,40 Interestingly, let-7a has been proved to be an ideal HCC therapy because cholesterol-conjugated let-7a (Chol-let-7a) was found to exhibit superior anti-HCC effects. 41 These effects promote tumor cell death, autophagy, and inhibit tumor cell growth, proliferation, invasion, and metastasis. Also, the re-introduction of miR-34a in HCC has been found to induce G1 arrest, apoptosis, and senescence and found to combat tumor growth, proliferation, scattering, migration, invasion, stemness, metastasis, chemo-resistance, and recurrence, and therefore it was identified as a potent HCC suppressor and an ideal therapeutic tool.3,27,29,42–47 Interestingly, miR-34a has been entered phase 1 clinical trial in HCC; 48 this is after significant tumor reduction, dramatic protection from progression without toxicity, and prolonged survival of animal models. 3 Besides, the interconnected functions between miR-34a and molecules of immune system in HCC have been verified through regulating the expression of chemokine CCL22 and recruitment of regulatory T (Treg) cells. 45 Also, it was found that the transfection of oncolytic adenovirus co-expressing miR-34a and interleukin 24 (IL-24) in HCC cells could effectively achieve tumor suppressive effects. 44 Furthermore, miR-34a has been demonstrated to sensitize apoptosis through CD95 network, which also provided substantial protective axis against the loss of let-7.49,50 Similarly, restoration of miR-199a expression in HCC cells has been shown to lead to 60% decrease of the invasive potential, inhibition of anchorage-independent growth, migration, proliferation, and cell cycle.32,34,51,52 In addition, miR-199a could sensitize HCC cells to doxorubicin and hypoxia-induced apoptosis. 32 Furthermore, miR-199 has been shown to function in immune system through regulating an important target in HCC, CD44 (a trans-membrane glycoprotein involved in cell–cell interaction, cell adhesion, and migration). 36
In the current study, we have demonstrated some important mechanisms of the most potent miRNAs regulating pathways of immune system and major HCC hallmarks through detecting miRNA-target genes and its regulated pathways. It has been shown that HCC is characterized by multiple cancer hallmarks.53–55 These hallmarks facilitated the elucidation of molecular pathogenesis of HCC and identification of effective therapeutics as well. 56 In this context, the aim of our current study is to perform an integrative in silico analysis for three potent tumor suppressor miRNAs—let-7a, miR-34a, and miR-199a/b—using a systematic bioinformatics approach. This is to investigate the synergy between these miRNAs on regulating specific network of protein-coding genes and pathways in HCC especially the concerted and novel ones that encompass cancer hallmarks and/or interactions with the components of the immune system. Supporting this perspective, we proceed on in vitro validating the inhibition of some novel oncogenes having critical roles in the induction and the progression of HCC. We highlight the role of some oncogenes such as SOX4 and we leave others for the next part of the study. Overall, these miRNAs may hold great promise as anti-HCC therapeutics.
Methods
We integrated different bioinformatics servers of miRNA-target prediction and functional enrichment analysis to get new insights into the multi-functions of three important tumor suppressor miRNAs—let-7a, miR-34a, and miR-199a/b—in immune system and different HCC/cancer hallmarks. Mainly, innovative servers (miRror Suite 57 and miRGator v3.0 58 ) were utilized to identify specific target genes from HCC expression data sets either for the combination of the miRNAs or for every individual miRNA. Then, the interactome between the miRNA and its corresponding targets in the enriched pathways and signaling interactions was determined by DAVID and GeneTrail. 59 The overall protocol of the study was presented in Figure 1.

Flowchart illustrating the integrative bioinformatics analysis steps of let-7a, miR-34a, and miR-199a/b in HCC.
The three potent tumor suppressor miRNAs—let-7a, miR-34a, and miR-199a/b—were analyzed by an integrative bioinformatics approach. First, the target genes which are regulated either by the combination of these miRNAs or by every individual miRNA were determined using miRror Suite or miRGator servers. The combinatorial target prediction analysis gave 87 common targets while the individual analysis gave 34 targets of let-7a, 36 targets of miR-34a, 40 targets of miR-199a, and 35 targets of miR-199b. Second, the interaction between the miRNA and its corresponding predetermined targets in a variety of the enriched pathways and Gene Ontology (GO) terms was revealed. This was done either by the global functional enrichment analysis of the common targets of the three miRNAs by the annotation tools (DAVID, REACTOME, STRING) or the individual functional enrichment analysis of every miRNA targets by GeneTrail (Kyoto Encyclopedia of Genes and Genomes (KEGG), GO, TRANSFAC, TRANSPATH, NIA human disease gene sets). Finally, the multi-functions of the three miRNAs of interest in pathways of immune system and major HCC/cancer hallmarks were identified.
Combinatorial analysis of miRNAs by miRror suite
The miRror v2.0 tool has been analyzed for the combinatorial regulation by let-7a, miR-34a, and miR-199a/b on common genes and pathways. 57 These miRNAs recognized common binding sites of targets, and therefore they exhibited overlapping roles. The analysis included two stages. (a) Target prediction of the common genes of the three miRNAs of interest based on the miRtegrate protocol. This protocol calculated the probability of matches between miRNAs and their target genes based on the statistical common coverage for all the miRNA-target prediction databases (DBs). These DBs were PITA TOP, 60 PicTar v5.0, 61 TargetScan, 62 microcosm, 63 microRNA.org, 64 DIANA-microT v3.0, 65 EIMMO-MirZ, 66 miRDB, 67 RNA22, 68 and MAMI (http://34.236.212.39/microrna/getMirnaForm.do). Our analysis focused on the highly expressed genes in HCC having top ranked score of prediction 25%. The significance threshold for determining the miRNA targets based on the hypergeometric distribution with correction for multiple testing is p ≤ 0.05. In addition, these predicted targets were filtered by miRror Internal Score (miRIS) which balanced between the miRror predictions and the DBs that support target prediction. We have rerun this analysis using miRIS range from 0 to 1. Additional step of analysis has been added which included testing the target validation with the three miRNAs using specialized DBs of target validation, for example, mirecords, mirtarbase, and tarbase. (b) Statistical functional enrichment analysis of the commonly determined targets in order to reveal the coordinated function and regulation by the three miRNAs. This enrichment has been evolved by forwarding the resulted common targets from miRror suite to several resources like (a) the DAVID functional annotation tool,69,70 for investigating the biological functions of the predetermined targets in pathways of KEGG, 71 BioCarta, and GO annotations. 72 (b) REACTOME, 73 a curated DB was implemented for additional pathway enrichment. Notably, for DAVID and REACTOME analyses, the significance was determined by the threshold (p ≤ 0.05) and multiple testing adjustments using false discovery rate (FDR). (c) STRING 74 DB was used to illustrate the possible protein–protein interaction network using cytoscape for the predicted targets of the miRNAs.
Individual analysis of miRNAs by miRGator v3.0
We used miRGator to predict the putative canonical targets with significant anti-correlation of expression with let-7a, miR-34a, and miR-199a/b in a portal of deep sequencing data of HCC from The Cancer Genome Atlas- Liver Hepatocellular Carcinoma (TCGA-LIHC).58,75 Accordingly, the analysis focused on the series of expression profiles of both miRNAs and mRNAs for the same set of samples to verify effective targeting relationship. Normalization between the data of the miRNA sequence and mRNA sequences was based on calculating the Pearson correlation coefficient and the Spearman rank correlation. Regarding the relationships between the three miRNAs of interest and their mRNA targets, our research model aimed to determine the common targets from the network viewer of at least two DBs of prediction and at most six. These DBs were PITA, 60 PicTar, 61 TargetScan, 62 microcosm, 63 miRDB, 67 and miRNA.org. 76 Mainly miRGator analyses depend on the highest opposite expression pattern between the miRNA and its targets which were closely placed. Consequently, a strong supporting evidence of targeting miRNA to these targets was proved. We have chosen the top 300 targets for each of these DBs. For network viewer of the miRNA–mRNA relation, the limited score ranged from 23 to 7 or 15 to 7 (score 23 or 15 showed the highest opposite expression pattern, while score 7 showed a decrease in the opposite expression pattern, and the targets were far placed from the miRNA). Interestingly, some of the predicted targets were verified in one or more DBs of target validation, for example, miRecords, 77 Tarbase, 78 and mirTarBase. 79 Consistently, we have run search in literature for all the determined miRGator targets to identify the novel ones and confirm the verified ones. Next, in order to reveal the enriched functional categories for these candidate targets, we forwarded them to GeneTrail server. 59
Functional enrichment analysis of miRNA targets by GeneTrail
We used GeneTrail to demonstrate the comprehensive regulation of the input lists of miRNA targets which were formerly determined from miRGator. This analysis covered various categories of immune system and/or major HCC/cancer hallmarks, and it based mainly on KEGG 71 and GO, 72 in addition to other interesting categories, for example, TRANSPATH 80 and TRANSFAC. 81 We performed Over-Representation Analysis (ORA) comparing a reference set of all genes to the input gene list of each miRNA (test set), and calculation of significance threshold (p ≤ 0.05) relied on hypergeometric distribution and multiple testing adjustment method: FDR.
In vitro validation
As mentioned above, target prediction analysis was performed to identify panels of novel oncogenic targets of the three miRNAs of interest. Consequently, we have proceeded on in vitro validation of some of the most important targets. We verified here one of them: SOX4 as a direct target of miR-34a.
Cell culture
The HCC cell line HepG2 was used as a cell culture model for the study, and it was obtained from American Tissue Culture Collection (ATCC, designation number HB-8065™). HepG2 was cultured in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% pen/strep under normal growth conditions (typically 37°C and 5% CO2).
In vitro transfection assay
HepG2 cells were seeded at a density of 4.0 × 105 cells/well in a six-well plate 24 h before transfection. The cells were further co-transfected with 6 µg of pEGP-miR-34a expression vector (DNA plasmid) using 12 µL of TurboFect in vitro transfection reagent according to the manufacturer’s recommendations (Thermo Fisher Scientific, Lithuania). After 4 h of incubation, the medium was replaced with fresh complete medium containing 10% FBS, and gene silencing was monitored 48 h post-transfection.
Quantitative validation of SOX4
Total RNA was extracted from the transfected and un-transfected HepG2 using BIZOL Reagent (BioFlux; Hangzhou Bioer Technology Co. Ltd., Binjiang) following the manufacturer’s instructions. Thereafter, complementary DNA (cDNA) was synthesized using the RevertAid™ H Minus First Strand cDNA synthesis kit as described by the manufacturer (Thermo Fisher Scientific) with oligo(dT)18 primers. Quantitative real-time polymerase chain reaction (qRT-PCR) for the expression of SOX4 transcript was performed using Universal SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) and specifically designed primer set: 5′-CAGCAAACCAACAATGCCGA-3′ and 5′-CTTGCACCAGCTCGGGTC-3′. All the reaction mixtures were added in 96 well plates and were put in Roche 480 thermal cycler under the following cycling conditions: 95 C for 10 min followed by 40 cycles of 95 C for 15 s, 50°C for 30 s, and 72°C for 30 s. For normalization of the expression level of SOX4, measurement of an endogenous control GAPDH mRNA was performed with the specific primer set: 5′-CAAGGTCATCCATGACAACTTG-3′ and 5′-GTCCACCACCCTGTTGCTGTAG-3′. The relative expression level was calculated by the formula 2−ΔΔCt. 82 The data were obtained from three independent experiments and expressed as mean ± standard error of the mean (SEM).
Results
Combinatorial targets of the miRNAs
There were 87 common predicted target mRNAs of the input miRNA set—let-7a, miR-34a, and miR-199a/b—including some novel ones (Table S1). These combinatorial targets resulted by the miRror suite DBs of target prediction and ranked according to p value with threshold 0.05. Afterward, the statistically significant and enriched pathways and GO terms of these targets have been determined by the DAVID functional annotation server and REACTOME. Overall, 12 enriched pathways have been identified. The collection of all these pathways was illustrated in Figure 2(a). Depending on our characterization of these pathways’ results, we found two pathways linked to immune system: regulation of innate and adaptive immune system pathways and cytokine–cytokine receptor interaction; we found eight pathways linked to HCC/cancer hallmarks: cellular responses to stress, cell cycle, signal transduction, gene expression, pathways in cancer, insulin signaling pathway, ERBB signaling pathway, and casitas B-lineage lymphoma (CBL)-mediated ligand-induced downregulation of epidermal growth factor (EGF) receptors; and we found two pathways that mediate interconnected dual function between immune system and major HCC/cancer hallmarks: focal adhesion and extracellular matrix signaling (ECM)-receptor interaction.

Bar chart of the enriched pathways and GO terms of the predicted targets of let-7a, miR-34a, and miR-199a/b, with the logarithm p value after FDR correction. (a) Pathways according to the determined significance thresholds (p ≤ 0.05), and pathway analysis of the common target genes by REACTOME, showing the highest six statistically significant pathways. While KEGG and BioCarta pathways analysis revealed four pathways and one pathway with lower significance. (b) GO functional categories of the commonly predicted targets of the miRNAs with the logarithm of corrected p value. According to the determined significance thresholds (p ≤ 0.05) and FDR adjustment, the GO analysis by DAVID tool revealed seven enriched GO terms of the common predicted targets.
Meanwhile, REACTOME provided us with some global high statistically significant (p < 0.001) pathway categories (Figure 2(a)), which are preferably divided into subcategories in order to clarify the functional enrichment of result. Examples of these pathways included signal transduction pathway category, which includes 13 subcategories: signaling by EGFR, signaling by FGFR, signaling by insulin receptor, signaling by PDGF, signaling by vascular endothelial growth factor (VEGF), signaling by ERBB2, signaling by ERBB4, PIP3 activating AKT signaling, RAF/MAP kinase cascade, signaling by TGF-beta receptor complex, signaling by Rho GTPases, signaling by NOTCH, and signaling by WNT. In addition, regulation of immune system pathway category includes innate immune system, adaptive immune system and cytokine signaling. Furthermore, gene expression category includes pathways of metabolism of non-coding RNA and miRNA-mediated RNA cleavage. And finally, pathways in cancer category which includes signaling by EGFR, PI3K/AKT signaling in cancer, signaling by FGFR, signaling by NOTCH1, signaling by TGF-beta receptor complex. As seen from the determined pathway results, let-7a, miR-34a, and miR-199a/b have interconnected functions on key immune system pathways besides other signaling pathways involved in HCC. These modulated functions were verified through the common predicted target genes of the three miRNAs in all these pathways. Examples of the most important predicted targets are shown in Table S2.
Regarding the result of DAVID GO annotations, there were seven enriched GO terms of the key predicted targets of let-7a, miR-34a, and miR-199a/b (Figure 2(b)) with p ≤ 0.05. Mostly, these GO categories were linked to HCC/cancer hallmarks for example, cell migration, cell proliferation and only one specific category; cell adhesion which was related to immunity with the highest significance (p-value 0.0081).
Individual targets of the miRNAs
We have determined a collection of common targets for each of the three miRNAs—let-7a, miR-34a, and miR-199a/b—from miRGator network viewer by at least two or at most six DBs of target prediction and score range from 23 to 7. Notably, all of the evolved targets had critical roles in hepatocarcinogenesis, and some of them have been considered to be novel as they were predicted by programs of target prediction but were not validated before. There were 34 predicted targets of let-7a, of which 13 genes of them were considered to be novel (Table S3). Meanwhile, there were 36 targets of miR-34a, in which nine genes of them were considered to be novel (Table S4). Also, there are 40 targets of miR-199a and 35 common targets of miR-199b. Interestingly, 22 genes of miR-199a and 25 genes of miR-199b were considered to be novel (Table S5 (A, B)). The miRNA-binding sites within the 3′ Un Translated Region (UTR) of some of these novel targets were shown in Figure 3(a)–(c). Importantly, we have narrowed the list of interesting novel targets and have chosen some of them for experimental validation. For example, SOX4 was validated as a direct target of miR-34a as it will be shown later. Furthermore, we could reveal the regulatory function of let-7a, miR-34a, and miR-199a/b in immune system modulation and/or HCC/cancer hallmarks through entering the target gene list of each of these miRNAs to GeneTrail.

The binding modes for the three miRNAs—let-7a, miR-34a, and miR-199a/b—and some of their novel targets. The predicted structure of the duplex between the miRNA and its target site (either 5′dominant canonical or 5′ dominant seed) was shown. (a) Representation of the mode of pairing between let-7a target sites within the 3′ UTR regions of FAM103A1 and DDX19B. (b) Representation of the mode of pairing between miR-34a target sites within the 3′ UTR regions of ACBD3 and PGAP2. (c) Representation of the mode of pairing between miR-199a/b target sites within the 3′ UTR regions of ABCA1 and ARHGAP21.
GeneTrail functional enrichment of let-7a
Overall, the 34-input target list of let-7a has yielded 15 significant KEGG pathways, 8 GO terms, and other interesting category: NIA human disease gene sets.
KEGG pathways
A total of 15 significant KEGG pathways were obtained; 3 pathways linked to immune system functions, 6 pathways linked to HCC/cancer hallmarks, and 6 pathways integrate interconnected networking between immune system and HCC hallmarks. These enriched pathways were tabulated in Table 1, where they are arranged in ascending order according to p value with threshold 0.05.
Enriched KEGG pathways for let-7a target genes.

KEGG: Kyoto Encyclopedia of Genes and Genomes; FDR: false discovery rate; HCC: hepatocellular carcinoma; MAPK: mitogen-activated protein kinase; VEGF: vascular endothelial growth factor.
GO terms
There were eight obtained GO terms in which the let-7a target genes had significant structural and molecular functions. These ontologies included nuclear pore, RNA localization, pore complex, nucleic acid transport, RNA transport, mRNA transport, and establishment of RNA localization. All these terms had unified significance of 0.0389916 except for nucleobase, nucleoside, nucleotide, and nucleic acid transport which had significance of 0.0491332.
Other significant category of let-7a included NIA human disease gene sets with one enriched item, neoplasms which suggested to be regulated by let-7a target genes with significance of 0.0335982.
GeneTrail functional enrichment of mir-34a
The 36-input target list of mir-34a has yielded two KEGG pathways, 22 significant GO terms, and other interesting categories: TRANSPATH and TRANSFAC.
KEGG pathways
There were two significantly enriched KEGG pathways where the mir-34a target genes have significant roles. These pathways were hematopoietic cell lineage and P53 signaling pathway with unified significance of 0.0454855.
GO terms
We have determined 22 GO terms in which mir-34a target genes have significant roles as shown in Table 2. These GO terms were arranged in ascending order according to their p value with threshold 0.05.
Enriched GO terms for miR-34a target genes.
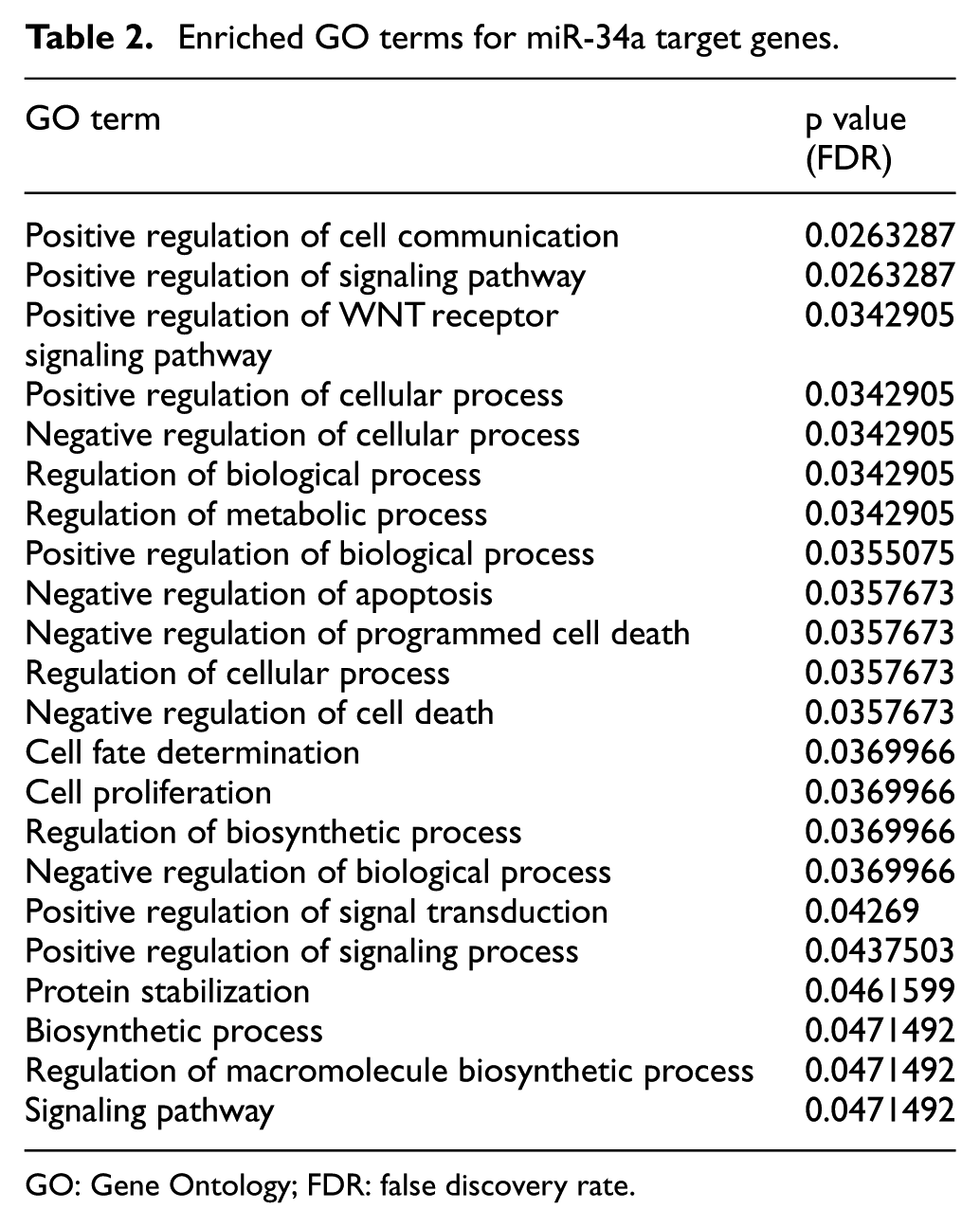
GO: Gene Ontology; FDR: false discovery rate.
Other categories of miR-34a enrichment analysis included TRANSPATH with one significantly enriched pathway which is NOTCH pathway (significance 0.00738002) and TRANSFAC with one significantly enriched function: Sp1 (significance 0.0421583).
GeneTrail functional enrichment of mir-199a/b
The 40-input target list of miR-199a has yielded four significant GO terms and other interesting TRANSPATH pathway.
GO term
We have determined four specific GO terms of the miR-199a target genes with significance of 0.0282994. The most important GO term was regulation of autophagy which provided new insight into a very specific functional role for miR-199a as a potent tumor suppressor in HCC. This could be demonstrated through miR-199a regulation of two important oncogenic targets, MTOR and CISD2, as determined from our findings. While the rest three terms had structural and molecular functions and they were intracellular part, membrane-bounded organelle and intracellular membrane-bounded organelle.
The other significant category of miR-199a included TRANSPATH with one enriched pathway which is ErbB3 survival with significance of 0.000444754.
The 35-input target list of miR-199b has yielded one KEGG pathway and eight significant GO terms.
KEGG category
Adipocytokine signaling pathway (p-value of 0.0368874) is the significantly enriched KEGG pathway in which the miR-199b target genes are implicated. In addition, adipocytokine signaling pathway was found to be significantly enriched according to our results with a p value of 0.0368874.
GO category
We have determined eight GO subcategories of the miR-199b target genes as shown in Table 3. These subcategories were arranged in ascending order according to p value with threshold 0.05.
Enriched GO terms for miR-199b target genes.

GO: Gene Ontology; FDR: false discovery rate.
In vitro validation results
SOX4 was validated as a direct novel target for miR-34a in the tested HepG2 cells by qRT-PCR analysis. After 48 h of pEGP-miR-34a transfection, the relative expression level of endogenous mRNA of SOX4 was significantly decreased by about 0.064% (15.6 folds) as compared to control (un-transfected cells, p < 0.05). In conclusion, this finding has shed light on the tumor suppression function of miR-34a in HCC through negative regulation of SOX4.
Discussion
The tumor suppression roles of let-7a, miR-34a, and miR-199a/b in HCC were reported in several studies.3,23,27,29,32,34,36,37,41,44–48,51,52 However, new functions of these miRNAs in pathways and GO categories of immune system and HCC hallmarks have been identified by our integrative analysis which is done by the bioinformatics tools; miRror Suite, DAVID, miRGator, and GeneTrail.
The enriched functions of the key common target genes by miRror
We have investigated the coordinated regulation of let-7a, miR-34a, and miR-199a/b on the targets with high statistical significance. Interestingly, examples of their novel fight against HCC included focal adhesion (represented interconnected link between immune system and HCC/cancer hallmarks) and insulin signaling (represented HCC/cancer hallmarks) (Figure 4(a) and (b)).

Two pathway phenotype examples showing the coordinated regulation of let-7a, miR-34a, and miR-199a/b in immune system functions and HCC/cancer hallmarks. The three miRNAs of interest let-7a, miR-34a, and miR-199a/b regulate common target genes in focal adhesion and insulin signaling. These targets promote a variety of hepatocarcinogenic phenotypes: (a) focal adhesion and (b) insulin signaling.
Focal adhesion
Aberrant expression and altered functions of focal adhesion proteins have been found to promote HCC progression, metastasis, and epithelial–mesenchymal transition (EMT) of hepatic cancer stem cells (CSCs).83,84 With reference to our result, focal adhesion was the highest statistically significant KEGG pathway (p value of 0.0077). It involved six predicted target genes (PAK4, FN1, PDGFRA, THBS1, CHAD, and ITGB8) as shown in Figure 4(a). PAK4 was found to be the highest significantly predicted target of let-7a, miR-34a, and miR-199a/b with p value of 0.0017. This oncogenic target has been reported to endogenously localize to adhesive structures and overexpress in HCC.85,86 Its activation has been shown to induce the loss of focal adhesion and promote HCC cells migration and metastatic invasion.86,87 Interestingly, it was reported that miR-199a/b target the tumor promoting PAK4 to suppress HCC growth, 21 while our findings revealed that the three miRNAs of interest suppress the oncogenic potential of PAK4 in focal adhesion and consequently inhibit worse immune response and hallmarks of HCC. FN1 is another significantly predicted target of let-7a and miR-199a/b found in our results with p value of 0.0029. Functionally, FN1 has been found to be involved in HCC cells survival and motility,88,89 and hence focal adhesion could be regulated through targeting FN1. Also, PDGFRA was found as a predicted target of let-7a, miR-34a, and miR-199a/b with p value of 0.0029. Interestingly, this is the first study to show the coordinated functioning of these three miRNAs in focal adhesion through regulating PDGFRA, which is known to induce HCC cells proliferation, migration, invasion, and EMT.90–92 Besides, these miRNAs were found to modulate cytokine–cytokine receptor interaction through the same interconnected target, PDGFRA. In addition, THBS1 was found as a predicted target of let-7a, miR-34a, and miR-199a/b with p value of 0.0038. It has been identified as a matricellular adhesive glycoprotein and angiogenesis stimulator in hepatocarcinogenesis. 93 Its overexpression has been shown to promote HCC cells’ adhesion, survival, proliferation, migration, and invasion.94,95 Herein, our study revealed that the three miRNAs efficiently block THBS1 and therefore act mainly as anti-angiogenic molecules. Collectively, these data implied that focal adhesion is a dual mediator between immune system and HCC hallmarks.
Insulin signaling
Insulin is strongly associated with glucose metabolism and cell proliferation, both of which are regulated by hepatocytes in physiological conditions.96,97 While, the HCC cells have been characterized by upregulated insulin signaling which leads to hyperinsulinemia and in turn promotes the survival and the proliferation of HCC cells.96,98 With reference to our result, insulin signaling was the second highest statistically significant KEGG pathway (p value of 0.0096). It involved five predicted target genes (MTOR, RHEB, CBL, CBLB, and PTPRF) as shown in Figure 4(b). MTOR was found to be the highest significantly predicted target of let-7a, miR-34a, and miR-199a/b with p value of 0.0001. Increased level and activity of this target have been found to be induced by insulin.99,100 Interestingly, this signaling has been demonstrated in 60% of HCC cases, which promoted cell cycle progression, proliferation, growth, and angiogenesis.101–105 Regarding the relation between let-7a, miR-34a, and miR-199a/b and MTOR as a direct target in insulin signaling, it has been shown that insulin stimulation inhibits the expression of miR-199a-5p. 106 Besides, miR-199a-3p targets MTOR in HCC cells, leading to G1 arrest, reduced invasive capability, and increased sensitivity to doxorubicin-induced apoptosis. 32 Moreover, it has been shown that let-7 together with LIN28A/B regulate the interaction between insulin and MTOR. 107 Loss of LIN28A and overexpression of let-7 resulted in insulin resistance due to repression of the insulin-MTOR signaling.107,108 Furthermore, miR-34a has been shown to interfere with MTOR signaling. 109 Collectively, based on these data, MTOR possibly acts as an important therapeutic target of let-7a, miR-34a, or miR-199a/b, and subsequently its contribution to previously mentioned HCC hallmarks will be suppressed. RHEB was found to be another significantly predicted target of the three miRNAs, and it is involved in insulin signaling with p value of 0.0023. This target has been known to be activated by its downstream target MTOR, and it is associated with cell cycle progression and growth.110,111RHEB/MTOR signaling is overactivated in a wide variety of cancers, emphasizing their oncogenic potential.112,113 Interestingly, insulin has been demonstrated to relieve inhibition of RHEB and increase its level which in turn activates the mTOR signaling.114,115 Therefore, based on the determined findings, the three miRNAs of interest have major inhibitory role on the multiconnected signaling of insulin/RHEB/MTOR.
LIN28B is among the key novel targets revealed by miRror prediction, and it provides a good example for the combinatorial HCC suppression by the three miRNAs of interest. This oncogene has been found to exhibit high expression in HCC with more frequency in high-grade HCCs. 116 In addition, it is well known to induce multiple oncogenic pathways in HCC, which promote tumor proliferation, EMT, and invasion capacity.116–118 Interestingly, LIN28B was reported to coordinate blocking let-7 precursors from being processed through terminal uridylation and degradation.117,119 Besides, our findings demonstrated that LIN28B is involved in regulating many of the non-coding RNA processes, including non-coding RNA/miRNA metabolic and catabolic processes, post-transcriptional regulation of gene expression, negative regulation of gene expression, gene silencing by RNA or miRNA, and production of miRNAs.
Regarding the protein–protein interaction network analysis of let-7a, miR-34a, and miR-199a/b targets which was retrieved from the STRING tool, 74 top ranking nodes of 24 interacting hub proteins (ATG4B, RHEB, TSC2, MTOR, EIF4EBP1, RICTOR, MLST8, RPTOR, RPS6KB1, AKT1, CBLB, CBL, PDGFRA, SPRY2, RANGAP1, RANBP2, RAD23B, ATAD2B, GRBS, MYEF2, SNAI1, MAP3K11, ACVR2B, and ACVR2A) were noticeably identified by cyto-hubba (Figure S1). By this interactome, the above-mentioned targets could link different pathways.
The enriched functions of the key common target genes by miRGator and GeneTrail
Individual target analysis for let-7a, miR-34a, or miR-199a/b has been conducted by miRGator platform which predicted the most prominent targets with inverse expression correlation to the miRNA of interest in HCC. Some of these targets were novel while others were validated. Our main focus was to reveal the vital regulatory roles of the three miRNAs on fine-tuning these targets. Importantly, we presented an integrative view for analyzing their novel mechanisms in some pathways and GO categories of the immune system and HCC hallmarks which have been determined by GeneTrail.
Let-7a and Toll-like receptor pathway
Normally, toll-like receptor (TLR) signaling has been known to be critical for regulating innate and adaptive immune responses in liver.120,121 However, abnormal TLR activation was found to develop inflammation-associated hepatocarcinogenesis due to their universal high expression.122–124 Our results revealed that let-7a regulation of TLR signaling is statistically significant (p value of 0.0110663) through two target genes (CASP8 and NFKB1). Notably, these targets were previously validated for let-7a;77,78 however, it is the first time to analyze their mediated action in TLR-driven HCC. Based on miRGator prediction analysis, CASP8 was a predicted target of let-7a with common score of 9.749 (Table S3). It has been found that CASP8 is highly expressed in the immune system and liver cells where it has novel regulatory role in inflammasome activation and induction of compensatory proliferation which are important for HCC progression.125,126 Thus, hepatocyte-specific deletion of CASP8 was found to be protective against inflammation-related hepatocarcinogenesis. 127 Consistently, our findings demonstrated direct targeting of CASP8 by let-7a, and consequently the HCC-promoting consequences of inflammatory responses (an emerging HCC hallmark; immune destruction) and multi-characteristics of HCC hallmarks are inhibited. Nuclear factor-kappa B1 (NF-κB1) was found to be another predicted target of let-7a with common score of 7.47 (Table S3). It has been previously shown that NF-κBs as well as TLRs are critical for the production of cytokines associated with hepatocarcinogenesis. 128 Actually, NF-κB activation was reported to be triggered by pro-inflammatory cytokines which produced through TLR signaling. 129 In addition, the activated NF-κB by TLR upregulation has been found to induce chronic pro-inflammatory response which transforms the liver stem/progenitor cells into HCC cells and promotes liver metastasis.130,131 Collectively, based on the accumulated evidence, let-7a can act as TLR signaling antagonist as it is shown previously to inhibit the immune destruction and other routine HCC hallmarks. Thus, it may represent an important antitumor tool against HCC.
Let-7a and NK cell–mediated cytotoxicity
NK cells have been reported to account for 25%–50% of liver lymphocytes and mediate natural cytotoxicity through producing cytokines and forming the first line of liver defense against infection, stress, and tumor formation.132,133 In HCC, NK cells have been found exclusively in areas of positive tumor apoptosis and negative tumor proliferation. 8 However, high percentage of these cells has been shown to be defective and might be associated with the immune evasion of HCC.134–137 Interestingly, let-7a was reported to be highly expressed in NK cells, 138 and according to our results, NK cell signaling is significantly regulated by let-7a (p value of 0.016632) through two predicted targets, NRAS and HRAS (Table S3). Although, these targets were previously validated for let-7a,41,77–79 it is the first time to investigate their actions in NK cell signaling in HCC. NRAS and HRAS have been shown to exhibit significant upregulation in hepatocarcinogenesis, and they regulate NK group 2 member D (NKG2D) ligand expression through mitogen-activated protein kinase (MAPK)/MEK and PI3K pathways.139–142 Therefore, direct targeting of RAS oncogenes by let-7a might provide defense mechanism against HCC because it acts as an enhancer for NK cell–mediated cytotoxicity and as an inhibitor for HCC-contributing pathways. Overall, NK cell signaling is essential as anti-HCC immunity. Thus, its augmentation may be a promising immunotherapy.
Mir-34a and positive regulation of WNT receptor signaling
The WNT receptor signaling is frequently activated in one third of all HCCs where it leads to limitless proliferation of tumor cells.1,143–145 With reference to our results, two enriched mir-34a target genes; SOX4 and PPM1A (p value of 0.0342905) were found to have roles in positive regulation of WNT receptor signaling in HCC. Based on miRGator analysis, SOX4 was found as a novel predicted target of miR-34a with common score of 7.226. SOX4 has been recognized as one of the most highly tumor-associated genes and cancer signature genes because it is significantly elevated in multiple cancers, including HCC.146–155 Besides, SOX4 was found to exert its oncogenic effect and metastasis-promoting capacity in stem cell compartment.156,157 We validated this target in HepG2 cells following transfection with pEGP-miR-34a vector. It exhibited downregulation by 15.6 folds as compared to control (un-transfected cells), respectively (p < 0.05). The fundamental role of SOX4 in positive regulation of WNT receptor signaling may be explained as follows: SOX4 has been demonstrated to regulate the transcriptional activity of many downstream target genes leading to HCC initiation, progression, EMT, and recurrence.150,158,159 For instance, SOX4 was showed to enhance WNT tumorigenic effects through direct interaction and stabilization of β-catenin/T-cell factor complex.150,151,160,161 These enhanced effects of SOX4 were associated with inhibition of apoptosis, cell cycle progression, proliferation, and inflammation.151,157,158,162 It has been found that RNA interference against SOX4 in invasive HCC greatly suppresses tumor growth, invasion, and metastasis.151,158 This finding supported our result which revealed that SOX4 is a potent target of miR-34a and its oncogenic role in WNT receptor signaling is reversed by this miRNA. PPM1A is a member of Ser/Thr protein phosphatases, 163 mainly expressed in the cytoplasm of HCC cells and so it is positively correlated with tumor progression. 164 PPM1A has been shown to dephosphorylate cyclin-dependent kinases to enhance cell invasion and migration.165,166 Our study revealed that PPM1A is a predicted and validated target of mir-34a, with common score of 7.0 (Table S4). PPM1A may enhance HCC cell growth through positive regulation of WNT receptor signaling. Nevertheless, further studies are still needed to investigate its clear mechanistic role. Collectively based on the mentioned data, we could conclude that mir-34a downregulates SOX4 and PPM1A which are involved in the sustained growth and proliferation of HCC cells through positive regulation of WNT receptor signaling.
Mir-34a and negative regulation of apoptosis
It has been shown that miR-34a induces apoptosis through regulating apoptosis inhibitors.43,167,168 Actually, miR-34a can participate in the apoptotic program triggered by p53 activation. 169 According to our results, five significantly enriched mir-34a target genes (NOTCH2, BIRC3, PGAP2, CD44, and SOX4) were found to act as apoptosis inhibitors in HCC (p value of 0.0357673). All these targets were predicted and validated for miR-34a except for SOX4 (Table S4). It has been shown that miR-34a inhibits NOTCH2 in order to promote apoptosis and inhibit proliferation and invasion of the tumor cells.170,171 However, our results showed that NOTCH2 is regulated by miR-34a in HCC cells to promote the same effects. BIRC3 has been known among the anti-apoptotic transcripts which downregulated by miR-34a leading to cancer cell death.172,173CD44 was identified as a molecular marker in multiple tumors including HCC because its aberrant overexpression has been correlated with the metastatic potential of tumors.174–177 In addition, it was shown to elevate in many CSCs including those of liver cancer and as a result it was identified as CSC marker evading death receptor–mediated apoptosis.178–184 Moreover, CD44 has been shown to promote apoptosis suppression in HCC to enhance adhesion-mediated proliferation, invasion, migration, and metastasis.174,185–187 Ectopic re-expression of miR-34a has been found to inhibit tumor cell growth and induce apoptosis 188 through downregulation of CD44. 189 In addition, miR-34a could abolish stemness of CSCs via direct targeting 90% of tumor-initiating CD44+ stem cells.175,190,191 Nevertheless, in the absence of miR-34a expression, CD44 has been shown to negatively regulate apoptosis via extracellular and intracellular signaling and mitochondrial pathway.192–196 Overall, our results confirmed the regulatory role of miR-34a on CD44 in HCC cells. As mentioned before, overexpression of SOX4 has been found to contribute to hepatic tumorigenesis by inhibiting p53-mediated apoptosis. 162 In this context, our findings provided additional evidence for the crucial role of miR-34a in positive regulation of p53-mediated apoptosis. Altogether, we could conclude that mir-34a opposes several HCC hallmarks such as apoptotic evasion, growth signaling, and proliferation through negative regulation of key oncogenic targets in apoptosis and consequently enhances HCC cell death.
MiR-34a and cell fate determination
With reference to our findings, two enriched mir-34a target genes were found to have roles in cell fate determination (p value of 0.0369966). These targets were NOTCH2 and JAG1 (validated for mir-34a); however, it is the first time to analyze their function in HCC cell fate determination. NOTCH signaling has been shown to play a pivotal role in cell fate decision, lineage commitment of lymphocytes, and defense against tumors. 197 However, its dysregulation has been shown to be implicated in tumor angiogenesis and metastasis. 198 NOTCH2 is an important receptor for membrane-bound ligands in NOTCH signaling. This receptor is involved in poor differentiation of many proliferating cells, including liver cells and self-renewal of stem cells. 199 Interestingly, NOTCH2 has been identified as HCC-associated oncogene, which exhibits high expression in neoplastic cells and hepatic CSCs.200,201 Liver-specific expression and oncogenic constitutive potential of NOTCH2 intracellular domain (N2ICD) haves been shown to activate NOTCH2 signaling and upregulate the pro-proliferative genes which stimulate the proliferation of hepatocytes and sustain the stemness of liver CSCs.200,202,203JAG1 was also a predicted target of miR-34a. It was shown to play an important role in cell fate determination through the JAG1-dependent NOTCH signaling, which is indispensable for stem cell self-renewal and differentiation. 204 Besides, this signaling is one of the major signals required for directing CSCs and promoting stemness in solid tumors.205–207 It has been reported that NOTCH2 and its ligand JAG1 is highly expressed in 83.3% of HCC tissues and their dysregulation leads to self-renewal of hepatic CSCs promoting hepatocarcinogenesis. 203 HCC gene expression profiling has confirmed the high expression of NOTCH2 and JAG1 as hepatic progenitor cell (HPC) markers. 208 Furthermore, it has been revealed that inhibiting either NOTCH2 or JAG1 could dramatically reduce tumor burden and growth. 202 Collectively, these results indicate that mir-34a suppresses the oncogenic potential of NOTCH2 and JAG1 which are key targets in HCC cell fate determination and consequently suppresses NOTCH signaling and its mediated HCC hallmarks.
Conclusion
In summary, this study highlighted the role of let-7a, mir-34a, and miR-199a/b as vital tumor suppressors in HCC. Our findings revealed that these miRNAs synergistically function together through interconnected regulatory networks consisting of common predicted target genes and enriched pathways of HCC. Also, each of these miRNAs individually regulates some specific targets and pathways. In addition, our results determined some novel regulatory actions of let-7a, mir-34a, and miR-199a/b in some pathways of immune system and HCC/cancer hallmarks (e.g. focal adhesion, insulin signaling, Toll-like receptor signaling, and adipocytokine signaling). Therefore, these miRNAs may act as effective inhibitors for HCC and may hold great promise either to be forwarded to preclinical and clinical trials or to be validated in other tumors.
Supplemental Material
supplementary_figure_TUB – Supplemental material for Bioinformatics functional analysis of let-7a, miR-34a, and miR-199a/b reveals novel insights into immune system pathways and cancer hallmarks for hepatocellular carcinoma
Supplemental material, supplementary_figure_TUB for Bioinformatics functional analysis of let-7a, miR-34a, and miR-199a/b reveals novel insights into immune system pathways and cancer hallmarks for hepatocellular carcinoma by Bangly Soliman, Ahmed Salem, Mohamed Ghazy, Nourhan Abu-Shahba and Mahmoud El Hefnawi in Tumor Biology
Supplemental Material
Supplementary_table(5)_forTUB – Supplemental material for Bioinformatics functional analysis of let-7a, miR-34a, and miR-199a/b reveals novel insights into immune system pathways and cancer hallmarks for hepatocellular carcinoma
Supplemental material, Supplementary_table(5)_forTUB for Bioinformatics functional analysis of let-7a, miR-34a, and miR-199a/b reveals novel insights into immune system pathways and cancer hallmarks for hepatocellular carcinoma by Bangly Soliman, Ahmed Salem, Mohamed Ghazy, Nourhan Abu-Shahba and Mahmoud El Hefnawi in Tumor Biology
Footnotes
Acknowledgements
Ahmed Salem, Mohamed Ghazy, and Mahmoud El Hefnawi conceived and supervised the study; Bangly Soliman and Mahmoud El Hefnawi designed the experiments; Bangly Soliman performed the in silico analysis, in vitro experiments, and the main statistical analysis; Bangly Soliman, Ahmed Salem, Mohamed Ghazy, and Mahmoud El Hefnawi analyzed the overall data; and Bangly Soliman and Mahmoud El Hefnawi were the major contributors in writing the manuscript, All authors have been involved in revising the manuscript, and all authors have given final approval of the version to be published. The authors are deeply indebted to Dr Ahmed Fawzy Mostafa, Associate Professor of Pharmaceutical Industries, National Research Center for his generous contribution in all practical experiments. Also, the authors are grateful to Engineer Saed Halim for helpful contribution in the preparation of figures and tables.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
Ethics approval and consent to participate for the current study were not required because the present study is mainly based on bioinformatics analysis.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Research Centre (grant no. P100106) and the Ministry of Higher Education and Scientific Research (Science and Technology Development Fund [STDF], grant no. 18505), Cairo, Egypt. These grants are awarded to Mahmoud El Hefnawi and Bangly Soliman, and the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.