
Abstract
Abnormal expression of long non-coding RNA often contributes to unrestricted growth of cancer cells. Long non-coding RNA XIST expression is upregulated in several cancers; however, its modulatory mechanisms have not been reported in hepatocellular carcinoma. In this study, we found that XIST expression was significantly increased in hepatocellular carcinoma tissues and cell lines. XIST promoted cell cycle progression from the G1 phase to the S phase and protected cells from apoptosis, which contributed to hepatocellular carcinoma cell growth. In addition, we revealed that there was reciprocal repression between XIST and miR-139-5p. PDK1 was identified as a direct target of miR-139-5p. We proposed that XIST was responsible for hepatocellular carcinoma cell proliferation, and XIST exerted its function through the miR-139-5p/PDK1 axis.
Introduction
Hepatocellular carcinoma (HCC), which ranks sixth in the list of most common tumors, currently represents the fastest growing cause of cancer-related deaths around the world. 1 Despite recent advances in HCC treatment, HCC still has a global mortality rate of 94%. 2 Therefore, it is necessary to develop novel strategies for the diagnosis and treatment of HCC.
Protein-coding genes account for only about 2% of the human genome, whereas the majority of transcripts consist of the non-coding RNAs (ncRNAs). The ncRNAs can be grouped into the following classes depending on their transcript size: long non-coding RNAs (lncRNAs) and small ncRNAs. 3 The lncRNAs are non-protein-coding transcripts with a length greater than 200 nt. A number of articles have suggested that lncRNAs are dysregulated in many cancers, including HCC. 4 Abnormal expression of lncRNAs often contributes to tumor initiation, metastasis, and cell growth.5,6 MicroRNAs (miRNAs), a class of small ncRNAs, usually bind to the 3′-untranslated region (3′-UTR) of messenger RNAs (mRNAs) which subsequently leads to mRNA degradation or translation repression. 7 It is well documented that alterations in miRNA expression play a critical role in cancer initiation and development. 8 Recent studies suggest that a potential function of lncRNAs is to competitively inhibit miRNAs as a molecular sponge. The interaction between lncRNA and miRNAs in cancer has been confirmed by several studies. For example, H19 lncRNA was shown to act as a molecular sponge for the let-7 family. However, let-7 could also negatively affect H19 expression. 9
In this study, we aim to identify the function of lncRNA XIST in promoting HCC cell growth. In addition, we investigate whether XIST affected the biological processes of HCC by regulating miRNAs.
Materials and methods
Patient samples and cell lines
A total of 88 HCC and matched-normal tissue samples were obtained from patients of the Gaozhou Hospital. Specimens were obtained during surgery and formalin-fixed and embedded in paraffin by standard methodologies. All HCC patients gave written consent to use the tissue samples for research purposes. This study was approved by the Ethical and Scientific Committees of the Gaozhou Hospital with the permitted number (GZRM-TU78). Human HCC cell lines, LM9, Hh7, Hep3B, and HepG2, and normal hepatic cell line LO2 were obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). All cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere in RPMI-1640 medium supplemented with 10% newborn bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For quantitative polymerase chain reaction (qPCR), 1 µg of total RNA was reverse transcribed to complementary DNA (cDNA) using a reverse transcription kit (Takara, Capitola Mall, Capitola, CA, USA). Real-time PCR analyses were conducted with Power SYBR Green (Takara). Results were normalized to the expression of U6 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers for XIST and miR-139-5p were previously described.10,11 The primers for XIST were 5′-CAGACGTGTGCTCTTC-3′ (forward) and 5′-CGATCTGTAAGTCCACCA-3′ (reverse). The primers for miR-139-5p were 5′- CTCCACTCCTCCCTTTCCTC-3′ (forward) and 5′-GCGGTAAGAAGCAGAGCAG-3′ (reverse). The primers for U6 were 5′-CTCGCTTCGGCAGCACA-3′ (forward) and 5′-AACGCTTCACGAATTTGCGT-3′ (reverse). The primers for GAPDH were 5′-AACGTGTCAGTGGTGGACCTG-3′ (forward) and 5′-AGTGGGTGTCGCTGTTGAAGT-3′ (reverse). The relative expression of each gene was calculated and normalized using the 2−ΔΔCt method relative to U6 small nuclear RNA (snRNA) or GAPDH.
Lentivirus production and transduction
Lentiviral small hairpin RNA (shRNA) targeting XIST was designed at http://biosettia.com/support/shrna-designer and cloned into the pLV-H1TetO-GFP-Puro vector according to manufacturer’s instructions (Biosettia Inc., San Diego, CA, USA). The viruses were packaged in 293T cells according to standard protocols, and the virus particles were harvested 72 h later. The packaged lentiviruses were named sh-XIST, and the empty lentiviral vector sh-ctrl was used as a control. HepG2 cells were infected with virus particles plus 8 µg/mL Polybrene.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, clonogenic assay, 5-ethynyl-2′-deoxyuridine assay, and apoptosis assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to assess cell proliferation was carried out as previously described. 12 Briefly, cells were seeded in 96-well plates. MTT (5 mg/mL) was added to each well, followed by an incubation of 4 h. After the supernatants were removed, dimethyl sulfoxide (DMSO) was added to each well and the absorbance value was measured at 490 nm.
For the clone formation assay, cells were seeded in six-well culture plates. After 14 days, the cells were stained with hematoxylin solution. After that, the number of colonies containing ⩾50 cells was counted.
The 5-ethynyl-2′-deoxyuridine (EdU) assay and apoptosis assay were performed as previously described. 13 For EdU assay, cells were seeded in 24-well plates, followed by incubation under standard conditions in complete media. Subsequently, the cells were incubated with 40 µM EdU for 2 h before fixation, permeabilization and EdU staining, which were performed according to the manufacturer’s protocol. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma, St. Louis, MO, USA) at a concentration of 1 µg/mL for 10 min. For apoptosis assay, cells were harvested and were resuspended in 1× binding buffer. After double staining with fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide (PI) using the FITC Annexin V Apoptosis Detection Kit I (BestBio, Shanghai, China), cells were analyzed using a FACScan® flow cytometer equipped with CellQuest software (BD Biosciences, San Jose, CA, USA) according to manufacturer’s instructions to detect early and late apoptosis of cells.
Oligonucleotide transfection
Synthesized RNA duplexes of miRNA control, miR-139-5p, and anti-miR-139-5p were obtained from RiboBio (Guangzhou, China). pcDNA-PDK1 was purchased from Biosettia Inc. Oligonucleotide transfection was performed with Lipofectamine 2000 reagent (Invitrogen) according to manufacturer’s instructions.
Western blot assay
Western blot assay was performed as previously described. 13 Briefly, equal amounts of protein were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked in 5% non-fat skim milk/Tris-buffered saline with Tween 20 (TBST), followed by incubation with primary antibodies at 4°C overnight. Subsequently, the membranes were incubated with appropriate secondary antibodies. The levels of goal protein were detected with enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA). The primary antibodies PDK1 (Lot: sc-30692; concentration used: 1:500), p-AKT (Lot: sc-271966; concentration used: 1:500), AKT (Lot: sc-1619; concentration used: 1:500), and GAPDH (Lot: sc-365062; concentration used: 1:1000) were purchased from Santa Cruz Biotechnology (Delaware Ave, Santa Cruz, CA, USA).
RNA-binding protein immunoprecipitation assay
RNA-binding protein immunoprecipitation (RIP) assay was performed using the EZ-Magna RIP Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instruction. Briefly, cells were lysed with the use of RIP lysis buffer, followed by an incubation step with RIP buffer containing magnetic beads conjugated with human anti-Ago2 antibody (Millipore) or negative control Normal Mouse IgG (Millipore). Proteinase K was used to digest the protein and then immunoprecipitated RNA was isolated. A NanoDrop spectrophotometer was used to measure the RNA concentration. Purified RNA was subjected to quantitative reverse transcription polymerase chain reaction (q-RT-PCR) analysis.
Phospho-kinase array and luciferase reporter assay
The human phospho-kinase array was purchased from R&D systems (McKinley Pl NE, Minneapolis, MN). The performance of the assay was carried out according to standard protocols.
For the luciferase reporter assay, the 3′-UTR untranslated region of PDK1 was amplified by PCR and cloned downstream of the firefly luciferase gene in the pGL3 vector (Promega, Kings Chase, Brentwood, UK). The vector was named wild-type (wt) 3′-UTR. Site-directed mutagenesis of the miR-139-5p binding site in PDK1 3′-UTR was performed using the Quick change site-directed mutagenesis kit (Stratagene, Cedar Creek, TX, USA) and named mutant (mut) 3′-UTR. Cells were transfected with wt-3′-UTR or mut-3′-UTR and miR-ctrl or miR-139-5p. The luciferase assay was performed using the dual luciferase reporter assay system (Promega) 48 h after transfection.
In vivo tumorigenesis
A number of 1 × 106 logarithmically growing cells (sh-ctrl and sh-XIST) were subcutaneously inoculated into the left–right symmetric flank of 6-week-old male BALB/c-nu/nu mice. At the end of the experiment, the mice were sacrificed and the harvested tumors were weighed and processed for Ki-67 staining.
Statistical analysis
SPSS 16.0 and GraphPad Prism 5.0 software were used for statistical analysis. Data are represented as mean ± standard error of the mean (SEM). Fisher’s exact test was used to identify differences between categorical variables. One-way analysis of variance (ANOVA) or two-tailed Student’s t-test was used for comparisons between groups. Differences were considered statistically significant when p < 0.05.
Results
XIST is upregulated in HCC tissues and cell lines and associated with tumor size
First, q-RT-PCR analysis was performed to determine the expression level of XIST in 88 pairs of human primary HCC tissue and their normal counterparts. It was found that XIST expression was significantly increased in the primary HCC tissues (Figure 1(a), p < 0.05). We further found that the HCC cell lines had higher levels of XIST expression than the normal hepatic cell line LO2 (Figure 1(b), p < 0.05). Subsequently, we investigated the potential associations between XIST expression and patients’ clinicopathological features. The clinicopathological features of HCC patients can be found in Table 1. Although XIST expression was not associated with parameters such as age (p = 0.119), gender (p = 0.754), alpha-fetoprotein (AFP) (p = 0.713), hepatitis B virus (HBV) (p = 0.383), and cirrhosis (p = 0.217) in HCC, high XIST expression was significantly correlated with tumor size (p = 0.002). Kaplan–Meier analysis revealed that high-level expression of XIST was associated with a shorter disease-free survival in patients with HCC (Figure 1(c), p < 0.05).
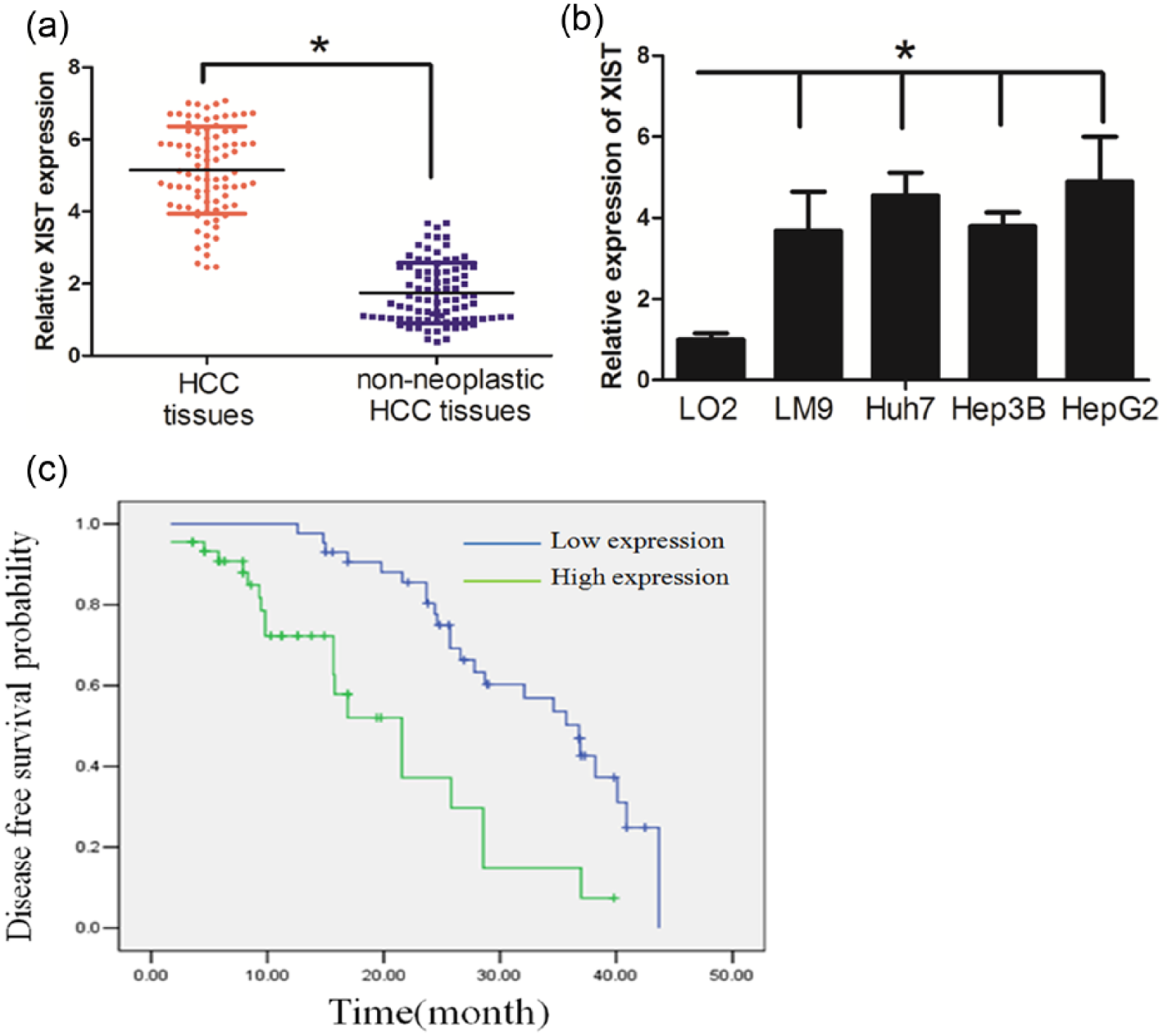
XIST expression was upregulated in HCC and associated with short disease-free survival of HCC patients: (a) XIST expression was significantly increased in the primary HCC tissues compared with normal counterparts, (b) HCC cell lines had higher levels of XIST expression than that in the normal hepatic cell line LO2, and (c) high-level expression of XIST was associated with short disease-free survival of patients with HCC (blue and green curves represent low- and high-expression of XIST, respectively; *p < 0.05).
Correction of XIST expression in tissue with patients’ clinicopathological features in HCC.

HBV: hepatitis B virus.
XIST inhibition decreased HCC cell proliferation and induced cell apoptosis
Since XIST expression was the highest in the HepG2 HCC cell line, we established HepG2 cells in which we stably knocked down XIST, and these cells were named sh-XIST. The cells treated with a negative control were named sh-ctrl (Figure 2(a)). The MTT assay revealed that knockdown of XIST significantly decreased cell proliferation in sh-XIST cells compared with sh-ctrl cells (Figure 2(b)). In parallel, the result of the colony formation assay demonstrated that oncogenic survival was significantly decreased in sh-XIST cells compared with sh-ctrl cells (Figure 2(c)).

XIST inhibition decreased HCC cell proliferation and induced cell apoptosis: (a) XIST expression in HepG2 transduced with control shRNA vector (sh-ctrl) or XIST shRNA vector (sh-XIST), (b) knockdown of XIST significantly decreased cell proliferation in sh-XIST cells compared with sh-ctrl cells, (c) colony formation assay demonstrated that oncogenic survival was significantly decreased in sh-XIST cells compared with sh-ctrl cells, (d) sh-XIST cells displayed a significantly higher frequency of cells at the G1 phase and a lower frequency of cells at S phase, (e) the number of cells incorporating EdU was significantly decreased in the sh-XIST group when compared with the sh-ctrl group, and (f) XIST downregulation could induce cell apoptosis.
Next, we performed flow cytometry analysis to examine whether XIST promoted HCC cell proliferation through altering cell cycle progression and/or apoptosis. The result showed that sh-XIST cells displayed a significantly higher frequency of cells at the G1 phase and a lower frequency of cells at the S phase (Figure 2(d)). This indicated that XIST promoted cell cycle progression from the G1 phase to the S phase. Similarly, the EdU incorporation assay revealed that the number of cells incorporating EdU was significantly decreased in the sh-XIST group when compared with the sh-ctrl group (Figure 2(e)). Interestingly, XIST downregulation could induce cell apoptosis in HCC (Figure 2(f)). Taken together, these data suggested that XIST promoted HCC cell proliferation by affecting the cell cycle and apoptosis.
Reciprocal repression between XIST and miR-139-5p in HCC
Several publications suggested that lncRNAs might participate in competing endogenous RNA (ceRNA) regulatory networks. We thus asked whether XIST exerted its function by regulating miRNAs expression and searched for miRNAs with complementary base paring with XIST utilizing the online software program starbase v2.0 (http://starbase.sysu.edu.cn/mirLncRNA.php). Because miR-139-5p was a tumor suppressor and was able to suppress cancer cell proliferation, we focused on miR-139-5p among these miRNAs that predicated may interact with XIST. Indeed, the RT-PCR assay showed that the miR-139-5p expression was increased in the sh-XIST group when compared with the sh-ctrl group (Figure 3(a)).

Reciprocal repression between XIST and miR-139-5p: (a) MiR-139-5p expression was increased in sh-XIST group compared with sh-ctrl group; (b) the binding sites of miR-139-5p on XIST; (c) co-transfection of miR-139-5p and XIST-Wt strongly decreased the luciferase activity, while co-transfection of miR-control and XIST-Wt did not change the luciferase activity, and co-transfection of miR-139-5p and XIST-Mut did not change the luciferase activity either; (d) MiR-139-5p negatively regulated XIST expression; and (e) XIST and miR-139-5p were enriched in Ago2 immunoprecipitates relative to control IgG immunoprecipitates.
Next, we asked whether miR-139-5p negatively regulated XIST expression. XIST was predicted to harbor miR-139-5p binding sites (Figure 3(b)). To further investigate whether XIST was a functional target of miR-139-5p, we cloned the predicted miR-139-5p binding site of XIST (XIST-Wt) and a mutated miR-139-5p seed region binding site of XIST (XIST-Mut) into a reporter plasmid. The results showed that co-transfection of miR-139-5p and XIST-Wt strongly decreased the luciferase activity, while co-transfection of miR-NC and XIST-Wt did not change the luciferase activity. Interestingly, co-transfection of miR-139-5p and XIST-Mut did not change the luciferase activity either (Figure 3(c), p < 0.05). We next assessed whether miR-139-5p was able to negatively regulate XIST expression. As shown in Figure 3(d), XIST expression was decreased in miR-139-5p groups, whereas the expression in the anti-miR-139-5p group was increased (p < 0.05). Taken together, these data suggested that miR-139-5p could directly bind to XIST and negatively regulated XIST expression.
It is documented that miRNAs exert their gene silencing functions through a ribonucleoprotein complex called the RNA-induced silencing complex (RISC). The core component of the RISC is Ago2. 14 RIP experiments were performed to determine whether XIST and miR-139-5p are in the same RISC complex. RT-PCR assay was used to determine RNA levels in immunoprecipitates. We found that XIST and miR-139-5p were enriched in Ago2 immunoprecipitates relative to control IgG immunoprecipitates (Figure 3(e), p < 0.05). These data indicated that both XIST and miR-139-5p are probably in the same RISC complex, consistent with our bioinformatic analysis and luciferase assays.
miR-139-5p/PDK-1 axis mediated the effect of XIST on cell growth in HCC
MiR-139-5p has been reported to inhibit cancer cell growth, and we asked whether the effect of XIST on cell growth was mediated by miR-139-5p. The MTT and colony formation assays showed that knockdown of XIST significantly inhibited the growth of HCC cells, while anti-miR-139-5p treatment rescued the effect (Supplementary Figure 1(A) and (B)). In addition, the alterations in cell cycle distribution and apoptosis, caused by XIST downregulation, were also rescued by anti-miR-139-5p treatment (Supplementary Figure 1(C) and (D)).
MicroRNAs have an influence on biological activity by regulating its downstream targets. 15 PDK1, a key regulator of cell growth in cancer development, 16 was identified as a downstream target of miR-139-5p (Figure 4(a)). Indeed, the luciferase assay showed that HCC cells co-transfected with miR-139-5p and wt-PDK1-3′-UTR had less luciferase activity than other groups (Figure 4(b)). The Western blot assay demonstrated that miR-139-5p repressed PDK1 protein expression in HCC cells (Figure 4(c)). In sh-XIST cells, we found that the level of PDK1 was less than that in sh-ctrl cells. However, anti-miR-139-5p treatment lead to the restoration of PDK1 in sh-XIST cells (Figure 4(d)). Interestingly, restoration of PDK1 rescued the effect the XIST knockdown had on cell growth, cell cycle distribution, and apoptosis (Supplementary Figure 2(A)-(D)). Taken together, these data suggest that the miR-139-5p/PDK1 axis mediated the effect of XIST on HCC cell growth.

miR-139-5p/PDK-1 axis mediated the effect of XIST on cell growth. (a) The binding sites of miR-139-5p on PDK1, (b) the luciferase assay showed that cells transfected with miR-139-5p had less luciferase activity than those transfected with miR-ctrl, (c) miR-139-5p repressed PDK1 protein expression in HCC cells, and (d) anti-miR-139-5p treatment lead to the restoration of PDK1 in sh-XIST cells.
XIST regulated AKT signaling in HCC through the miR-139-5p/PDK1 axis
Receptor tyrosine kinases (RTKs) are documented to play crucial roles in HCC proliferation; we thus asked whether XIST could affect RTK signaling pathways in HCC. A human p-RTK array was used to detect the tyrosine phosphorylation level of 42 different RTKs.17,18 We found that phosphorylation of protein kinase B (Akt-T308) was significantly decreased in sh-XIST cells (Figure 5(a)). The Western blot assay was consistent with the p-RTK array (Figure 5(b)). PDK1 is well known to phosphorylate AKT at T308, 19 and we asked whether XIST regulated the AKT signaling pathway through the miR-139-5p/PDK1 axis. It was revealed that anti-miR-139-5p treatment or overexpression of PDK1 could rescue the phosphorylated AKT expression level in sh-XIST cells (Figure 5(c)). These data suggest that XIST regulated AKT signaling in HCC through the miR-139-5p/PDK1 axis.

PDK1 regulated p-AKT expression through miR-139-5p/PDK1 axis: (a) Akt-T308 was significantly decreased in sh-XIST cells, compared with that in sh-ctrl cells, (b) the Western blot assay revealed that XIST downregulation decreased p-AKT expression, and (c) anti-miR-139-5p treatment or overexpression of PDK1 could rescue the phosphorylate AKT expression level in sh-XIST cells.
XIST inhibition decreased tumor growth in vivo
We investigated whether knockdown of XIST could inhibit tumor growth in vivo. Compared with sh-ctrl cell-derived xenograft tumors, sh-XIST cell-derived xenograft tumors grew more slowly (Figure 6(a)). The mean weight of sh-TUG-1 cell-derived xenograft tumors was also significantly less as compared with sh-ctrl cell-derived xenograft tumors (Figure 6(b)). Then, the tumor sections were stained for Ki-67 expression to quantitatively assess the proliferation index in xenograft tumors (Figure 6(c)). Taken together, these data support the growth-promoting effect of XIST in vivo.

XIST inhibition decreased tumor growth in vivo: (a) compared with sh-ctrl cell-derived xenograft tumors, sh-XIST cell-derived xenograft tumors grew more slowly; (b) the mean weight of sh-TUG-1 cell-derived xenograft tumors was also significantly less as compared with sh-ctrl cell-derived xenograft tumors; and (c) knockdown of XIST significantly decreased the percentage of Ki-67 positive cells in tumors as compared with the negative control group.
Discussion
Evidence has shown that lncRNAs may serve as effective therapeutic targets for cancer treatment, including breast cancer, prostate cancer, colon cancer, and HCC. 20 However, only a few lncRNAs have been functionally characterized. In a previous study, we identified several lncRNAs which can be considered as a potential prognostic indicator for HCC. 21 Dysfunctional expression of XIST may have a pathological role in cancer. LncRNA XIST can promote cancer cell proliferation and invasion in glioma. 22 In addition, lncRNA XIST can be a predictive biomarker for screening non–small cell lung cancer. 23 Abnormal expression of lncRNA NEAT1 has a close relationship to the development of ovarian cancer occurrence, growth, invasion, and metastasis. 24 In this study, we revealed that XIST was upregulated in HCC patient tissues and cell lines. Interestingly, high XIST expression was significantly correlated with tumor size. This result stimulated us to investigate the underlying mechanism of XIST in regulating HCC cell growth. The functional study revealed that knockdown of XIST decreased cell proliferation both in vitro and in vivo. Further investigation found that XIST promoted cell cycle progression from the G1 phase to the S phase. In addition, XIST may protect HCC cells from apoptosis. These data suggest that XIST functions as an oncogene and drives carcinogenesis by promoting cell proliferation in HCC.
Emerging evidence suggests that lncRNAs act as endogenous miRNA sponges to bind to miRNAs and regulate their function.25,26 To find out whether XIST served as a miRNA sponge, we performed bioinformatic analysis and found that XIST contained binding sites for several miRNAs. We focused on miR-139-5p, which has been reported to regulate cell proliferation in cancer.27,28 A luciferase assay indicated that miR-139-5p could bind to XIST directly by the putative miRNA response element. Furthermore, overexpression of miR-139-5p suppressed XIST expression, whereas downregulated miR-139-5p induced a reverse result. Interestingly, a XIST knockdown displayed elevated miR-139-5p expression. The above data suggest that there might be a reciprocal repression between XIST and miR-139-5p. Finally, we found that XIST and miR-139-5p were in the same RISC complex by RIP assays, suggesting that there was a physical interaction in HCC cells. We further investigated whether miR-139-5p mediated the tumor-suppressive effect of XIST knockdown in HCC. Our present data indicated that while the knockdown of XIST decreased HCC cell proliferation, the inhibition of miR-139-5p could rescue the effects that the knockdown of XIST exerted.
PDK1 was identified as a potential target of miR-139-5p in our study and miR-139-5p negatively regulated PDK1 expression in HCC cells. As a constitutively active phospholipid-binding kinase, PDK1 can activate AKT kinase on the activation loop at T308, which promotes full activity of AKT. 29 In addition, the constitutive activity of PDK1 is a critical regulator of several other important signal transduction pathways that regulate cell proliferation, survival, and apoptosis. 30 Activation of PDK1 upregulated cyclin D1 during cell cycle progression from G0–G1 to S phase. 9 In this study, we found that XIST positively regulated PDK1 expression through miR-139-5p inhibition. Interestingly, restoration of PDK1 rescued the effect of the XIST knockdown on cell growth, cell cycle distribution, and apoptosis. These data demonstrated that XIST may exert its function through the miR-139-5p/PDK1 axis.
PDK1 is a transducer of PI3K signaling and activates multiple downstream effectors. 10 AKT can be phosphorylated by PDK1 at T308. With the use of a human p-RTK array, we found that XIST knockdown led to decreased phosphorylation expression of AKT-T308. Anti-miR-139-5p treatment or overexpression of PDK1 could rescue the phosphorylate AKT expression level in sh-XIST cells. Inhibition of AKT activity suppressed cancer cell proliferation by decreasing expression of CycD1 and Ki-67. 11 In addition, block of AKT activity could induce cancer cell apoptosis through activating caspase-3/9 while inhibiting bcl-2. 12 These literatures suggest that AKT activation play key role in regulating cancer cell proliferation and apoptosis. We proposed that XIST promoted cell growth by regulating the AKT signaling through miR-139-5p/PDK1 axis in HCC.
In summary, we demonstrated that XIST was upregulated in HCC tissues and was associated with worse survival of HCC patients. In addition, we uncovered that XIST activated AKT signaling through miR-139-5p/PDK1 axis. Our findings might facilitate the development of lncRNA-directed diagnostics and therapeutics against HCC.
Footnotes
Acknowledgements
Y.M., Y.L., P.W., S.H., and L.H. contributed equally to this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.