
Abstract
Morbidity in advanced prostate cancer patients is largely associated with bone metastatic events. The development of novel therapeutic strategies is imperative in order to effectively treat this incurable stage of the malignancy. In this context, Akt signaling pathway represents a promising therapeutic target able to counteract biochemical recurrence and metastatic progression in prostate cancer. We explored the therapeutic potential of a novel dual PI3 K/mTOR inhibitor, X480, to inhibit tumor growth and bone colonization using different in vivo prostate cancer models including the subcutaneous injection of aggressive and bone metastatic (PC3) and non-bone metastatic (22rv1) cell lines and preclinical models known to generate bone lesions. We observed that X480 both inhibited the primary growth of subcutaneous tumors generated by PC3 and 22rv1 cells and reduced bone spreading of PCb2, a high osteotropic PC3 cell derivative. In metastatic bone, X480 inhibited significantly the growth and osteolytic activity of PC3 cells as observed by intratibial injection model. X480 also increased the bone disease-free survival compared to untreated animals. In vitro experiments demonstrated that X480 was effective in counteracting osteoclastogenesis whereas it stimulated osteoblast activity. Our report provides novel information on the potential activity of PI3 K/Akt inhibitors on the formation and progression of prostate cancer bone metastases and supports a biological rationale for the use of these inhibitors in castrate-resistant prostate cancer patients at high risk of developing clinically evident bone lesions.
Introduction
Prostate cancer (PCa) continues to be a leading cause of cancer death in industrialized countries among male population. 1 Therapeutic strategies, blocking androgen receptor (AR), have been used for the management of locally advanced or advanced PCa 2 and are initially effective inducing a mixed response of cell cycle arrest and apoptosis. Progressively, patients with PCa develop castration-resistant disease (CRPCa) with bone metastatic disease and chemotherapeutic resistance. 3 Tumor progression and metastasis are complex processes with alterations at both genetic and epigenetic levels. Metastatic carcinoma cells degrade the extracellular matrix (ECM) and escape the site of the primary tumor migrating through blood or lymphatic circulation and colonizing remote sites including the bone marrow. 4 These processes are associated to epithelial–mesenchymal transition (EMT), a reversible cellular reprogramming process which induces changes in cell shape and initiates carcinoma dissemination.5,6 Important players for EMT are transforming growth factor-β (TGF-β) and CC chemokine ligand 2 (CCL2)/monocyte chemoattractant protein 1 (MCP-1)7–10 able to sustain intracellular PI3K/Akt activity. 11 mTOR is a serine/threonine protein kinase downstream to Akt that interacts with several proteins to mTORC1 and mTORC2 complexes. mTORC1, containing Raptor, is sensitive to rapamycin and controls protein synthesis, translation initiation, and cell mass through the phosphorylation of the ribosomal S6 kinase (S6K) and the elongation factor binding protein (4E-BP1). MCP-1 is able also to inhibit adenosine monophosphate-activated protein kinase (AMPK), a negative modulator of Akt, and this mechanism is necessary to sustain mTORC1 activation and p70S6K phosphorylation as well as upregulation of survivin and promotion of cell survival. 12 Conversely, mTORC2 regulates cell survival and proliferation,13–15 chemosensitivity,16,17 and radiosensitivity.18,19 Rictor knockdown arrests tumor cells in an intermediate stage between epithelial and mesenchymal differentiation, without the motile and invasive behavior of cells. 20 Raptor and rictor levels are elevated in advanced PCa cases with a predictive progression value 14 and modulating tumor dormancy in metastatic sites. 21 mTORC2 is required for a complete TGF-β-mediated EMT.11,13,21–23 In addition, mTORC1 is important for osteoblast and osteoclast function.24–26 It has been also demonstrated that PCa cells produce soluble factors accelerating osteoclast 27 and/or osteoblast differentiation. 28 For these reasons, mTORC2 might be an effective target for the prevention of cancer metastasis.29,30 The development of a tumor proliferation niche in the bone marrow31,32 is considered as the effect of resident or recruited stromal cells, bone marrow–derived cells and signals, or secreted factors including cytokines, chemokines (SDF1a), 33 and exosomes. 34 In addition, the presence of an immune infiltrate is essential for tumor progression, 35 since treatment with the broad-spectrum anti-inflammatory drugs protects against PCa formation and progression.36–38 Neutrophils, the first responders of acute tissue damage, have been associated with cancer progression and metastases. 39 The identification of macrophages as key contributors to tumor formation and progression is consistent with studies in a wide variety of other tumor types40,41 including PCa. Non-selective inhibitors of signaling pathways downstream PI3K/mTOR have demonstrated potent antiproliferative and pro-apoptotic effects in several human PCa xenograft models.42,43 Dual targeting of PI3K and mTOR could represent an effective strategy in order to prevent the activation of alternative pathways, to block with increased efficiency a complex intracellular oncogenic signaling network. Here, we explored the effect of the dual PI3K/mTOR inhibitor, X480, on the bone metastatic ability, osteoclast activation, and osteoblast function of PCa cells in vitro and in vivo. Our results demonstrate that X480 is able to decrease the metastatic potential of PCa cells through a pleiotropic antimetastatic activity that includes the inhibition of several steps during metastatic spreading from distant seeding to bone cell interaction. So, X480 administration could be a useful therapeutic approach in CRPCa patients at high risk of developing bone metastatic lesions.
Materials and methods
CRPCa cell models
22rv1 (DSMZ, Frankfurt, Germany), VCaP (REF), LnCaP, PC3, and DU145 (ATCC, LGC standards, Teddington, UK), C4-2B, PC3 lung, and PCb2 44 were used for in vitro experiments. Non-tumor EPN and BPH-1 cells were used as the control cells. PC3-M stably transfected cells with luciferase were kindly provided by Gabri van der Pluijm (Leiden University, The Netherlands). THP-1 tumor-associated macrophages were kindly provided by Emanuela Corsini (University of Milan, Italy). Mouse monocytic cell line RAW 264.7 was kindly provided by Prof E Tolosano (University of Torino, Italy). Osteoblast-like MC3T3-E1 cells were kindly provided by Dr G Tulipano (University of Brescia, Italy). Cells were grown as described in the original reports.
Reagents
All the materials for tissue culture were purchased from HyClone (Cramlington, NE, USA). Anti-total Akt (Sc-377457), p-Akt Thr308 (sc-16646R/sc-135650), p-Akt Ser473 (sc-135651), p-mTOR (Ser 2448), total mTOR (N19, SC-1549), rictor (H11, sc-271081), and raptor (10E10, sc-81537) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). In-Cell ELISA (also known as cell-based enzyme-linked immunosorbent assay (ELISA), In-Cell western or cytoblot assay) was used to quantify target proteins in cultured cells. For this analysis, the abovementioned antibodies are used following the “In-Cell ELISA protocol” available at http://www.abcam.com/protocols/in-cell-elisa-ice. Immunohistochemical analyses were performed on tissue arrays from primary prostate cancer tumors and bone metastases from US Biomax (Rockville, MD, USA) though European distributor (Gentaur, Brussels, Belgium).
Drug preparation
X480 (Xcovery FLN, Palm Beach Gardens, FL, USA) was stored as a dimethyl sulfoxide (DMSO) dissolved powder at 4° C and suspended in the medium on the day of use. A stock solution of drugs was prepared. In vivo, we used a 5% polyethylene glycol 400 (PEG-400), 5% ethanol, and 2% Tween-80 in a purified water solution to dissolve X480 administered at 3 mg/kg every day.
Osteoclastogenesis and osteoblastogenesis assays
RAW 264.7 cells were incubated with conditioned media collected from cells untreated or treated with X480 PCa in the presence or not of 5 ng/mL receptor activator of nuclear factor kappa-β ligand (RANKL). Tartrate-resistant acid phosphatase (TRAP) staining was determined by Leukocyte Acid Phosphatase Assay kit (Sigma-Aldrich, St Louis, MO, USA). Cells were washed and TRAP-positive multinucleated cells containing three or more nuclei were counted using a light microscope. For osteoblastogenesis experiments, we used three experimental models: osteoblast-like MC3 T3-E1, bone marrow stromal cells, and osteoblasts from calvaria representing different phenotypes of murine osteoblasts. Bone marrow stromal cells and osteoblasts from calvaria were isolated as previously described.44,45 Osteo-derived cells were seeded onto 24-well culture dishes and then allowed to grow to confluence. Conditioned media from untreated and X480-treated PC3 and C4-2B cells were used at 30% in the presence of α-modified eagle medium (αMEM) containing 10% fetal calf serum (FCS) or mineralized medium (αMEM containing 10% FCS, 100 µg/mL ascorbic acid, and 5 mM glycerol-2-phosphate) for an additional 7–21 days. Osteoblast phenotype was evaluated by cytochemical analyses of alkaline phosphatase (ALP) activity kit (Sigma-Aldrich, Milan, Italy). An indirect quantification of osteoblasts was also used by Alizarin Red S Staining Quantification Assay (ARed-Q; ScienCell, Carlsbad, CA, USA). Osteoblast differentiation was also evaluated in semi-quantitative polymerase chain reaction (PCR) assay using primers for collagen 1a2 and osteonectin.
Pharmacokinetic analysis
Pharmacokinetic (PK) analysis was performed by Sundia MediTech, Shanghai, China. Briefly, 12 mice were randomly divided into two groups receiving X480 (5% ethanol, 5% PEG-400, and 2% Tween-80 in physiological saline) administered at 3 and 10 mg/kg by oral gavage daily for 11 days. Blood samples (0.5–1.0 mL) were collected at different time points and stored on ice prior to centrifugation. The samples (within 15 min after blood collection) were centrifuged at 3000 rev/min for 10 min and the plasma samples were stored at approximately −20°C until being analyzed by liquid chromatography with tandem mass spectrometry (LC–MS/MS).
Xenograft model
Male CD1 nude mice (Charles River, Milan, Italy) were maintained according to the guidelines established by our Institution (University of L’Aquila, Medical School and Science and Technology School Board Regulations, complying with the Italian government regulation n.116 January 27 1992 for the use of laboratory animals). All mice received subcutaneous flank injections of 1 × 106 PC3 or 22v1 cells. Tumor growth was assessed biweekly by measuring tumor diameter with a vernier caliper (length × width). Tumor weight was calculated according to the formula: tumor volume (mm3) = 4/3πR1 × R2 × R3 as previously described. 16 Animals were sacrificed by carbon dioxide inhalation and tumors were subsequently removed surgically. Indirect immunoperoxidase staining of tumor xenograft samples was performed on paraffin-embedded tissue sections (4 µm) following standard conditions.
Intracardiac tumor model
Heart injection of PCa cells was performed as previously described. 46 Briefly, a 27-gauge needle on a tuberculin syringe containing 1 × 105 tumor cells in 0.1 mL of phosphate-buffered saline (PBS) was inserted in the second left intercostal space of male CD1 nu/nu mice. The development of metastases was monitored by radiography using a Faxitron cabinet X-ray system (Faxitron X-ray corp., Wheeling, IL, USA). Burden of osteolytic lesions was quantified by image analysis using ImageJ 1.43r (Wayne Rasband, National Institutes of Health (NIH)). Animals were sacrificed by carbon dioxide inhalation 170 days after heart injections, or earlier if there were early signs of serious distress. All animals were subjected to an accurate necroscopy and portions of various organs were processed for routine histological examination.
Intratibial tumor model
Intratibial tumor injection was performed as previously described. 46 Briefly, luciferase-transfected PC3Luc cells were injected at 2 × 105 cells/10 µL of serum-free medium into the proximal tibiae of 4-week-old male CD1 nu/nu mice. The development of metastases was monitored by radiography using a Faxitron cabinet X-ray system (Faxitron X-ray corp.) and tumor burden was evaluated by bioluminescence analyses (see below).
Treatments of bone metastatic models
Before drug administration, the animals were randomized into two groups of treatment as follows: Group 1—mice (10 animals) receiving 100 µL vehicle postoperatively (p.o.); Group 2—mice (10 animals) receiving X480 (100 µL, 3 mg/kg every day p.o.).
Assessment of treatment response in bone metastatic models
Tumor and bone treatment response was determined using X-rays as well as histomorphometric analysis of H&E-stained sections. Skeletal lesions were calculated as described by Yang et al., 47 where 0 = no X-ray-detectable lesions, 1 = minor lesions, 2 = small lesions, 3 = significant lesions with minor break of margins, or 4 = significant lesions with major break of margins.
Bioluminescence analyses
For bioluminescence imaging, the mice received 150 mg/kg intraperitoneal (i.p.) firefly luciferase (Synchem, Felsberg-Altenburg, Germany). Following anesthesia with ketamine/xylazine mixture, the mice were placed into a Hamamatsu imaging station (Hamamatsu photonics, Rome, Italy). Bioluminescence generated by the luciferin/luciferase reaction was used for quantification using a dedicated Living Image software.
Micro-computed tomography (μCT) analysis
After the animals were sacrificed, tibias and femurs were fixed in 4% formaldehyde and acquired in a SkyScan 1174 with a voxel size of 6 μm (X-ray voltage: 50 kV). The scans were over 180 degrees with a 0.3-degree rotation step. Image reconstruction was carried out employing a modified Feldkamp algorithm using the SkyScan NRecon software.
Serum levels of C-telopeptide of type I collagen and TRAP5b
Measurement of serum mouse cross-linked C-telopeptide of type I collagen (C-telopeptide of type I collagen (CTX-1), USCN Life Science Inc., Houston, TX, USA) and mouse tartrate resistant acid hosphatase (mTRAP5b) (MyBioSource, Inc., San Diego, CA, USA) were performed according to the manufacturer’s protocol.
Statistical analysis
Continuous variables were summarized as mean and standard deviation (S.D.) or 95% confidence interval (CI) for the mean. Statistical comparisons between the control and treated groups were established by carrying out the analysis of variance (ANOVA) test or by Student’s t test for unpaired data (for two comparisons). Dichotomous variables were summarized by absolute and/or relative frequencies. For dichotomous variables, statistical comparisons between the control and treated groups were established by carrying out the Fisher’s exact test. For multiple comparisons, the level of significance was corrected by multiplying the P value by the number of comparisons performed (n) according to Bonferroni correction. For matched pairwise multiple comparisons, dichotomous variables were compared with Cochran’s Q test. For matched pairwise comparisons, McNemar’s test was used. Overall and disease-free survival were analyzed by Kaplan–Meier curves and Gehan’s generalized Wilcoxon test. When more than two survival curves were compared, the log-rank test for trend was used. This tests the probability that there is a trend in survival scores across the groups. All tests were two-sided and were determined by Monte Carlo significance. P values < 0.05 were considered statistically significant. MedCalc software package (MedCalc, Ostend, Belgium) was used for statistical analysis and graphic presentation.
Results
Expression of p-Akt (Ser473), p-Akt (Thr308), and p-mTOR (Ser 2481 55.42) in PCa cell models and primary and metastatic human prostate tissues
First, we analyzed by cell-cell based ELISA the expression levels of Akt and mTOR in phosphatase and tensin homolog (PTEN)-positive (DU145, 22rv1, and VCaP) and PTEN-negative (LnCaP, C4-2B, PC3, and PC3 cell derivatives such as PCb2, PC3Lung). VCaP, C4-2B, PC3, and PCb2 cells represent cell models with higher bone tropism.
We demonstrated, in agreement with previous observations, that higher levels of p-Akt (Thr308), p-Akt (Ser473), and p-mTOR (Ser2448) were observed in PTEN-negative compared to PTEN-positive cells (Figure 1(a)) More aggressive and bone metastatic PCa cells also possessed higher levels of mTOR activity. To further demonstrate the upregulation of Akt/mTOR pathways in advanced and metastatic PCa tissues p-Ser473 and p-Thr308 Akt, p-mTOR, rictor, and raptor were immunohistochemically evaluated in PCa human tissue arrays (Gentaur, Brussels, Belgium).
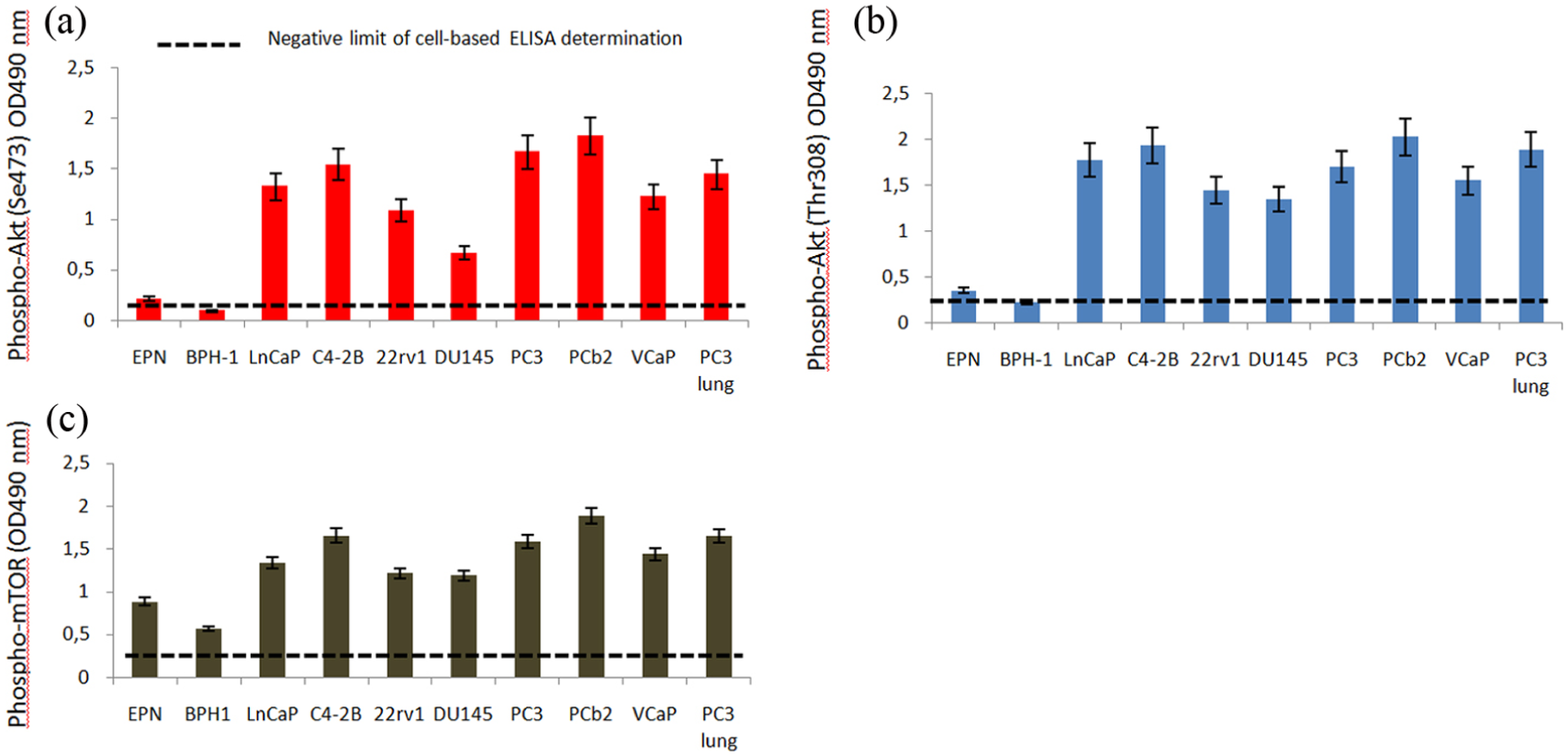
Expression of p-Akt (Ser473), p-Akt (Thr308), and p-mTOR (Ser 2481 55.42) in PCa cell models: (a) ccell based ELISA for p-Akt (ser473); (b) ccell based ELISA for p-Akt (Thr308); and (c) ccell based ELISA for p-mTOR (ser2448). Data are representative of an individual experiment performed in quintuplicate.
We observed, indeed, that the activation of Akt and mTOR was higher in high Gleason grade scores when compared to those observed in low Gleason grade PCa (Gleason score < 7) and in bone metastatic sites when compared to primary tumors (Figure 2). Similarly, raptors were higher in primary tumors and rictors in bone metastases.

Immunohistochemical evaluation of expression levels of members of Akt/mTOR pathways in human tissue samples: (a) representative pictures showing immunohistochemical expression pattern versus p-Akt (Ser473), p-Akt (Thr308), p-mTOR, rictor, and raptor. (b) Comparisons between BPH and PCa with Gleason scores ⩽7 and >7. Immunohistological score 2 + was considered as high expression. (c) Representative picture for p-mTOR in 22rv1, DU145, and PC3 subcutaneous xenografts and PC3 bone metastases.
In vitro characterization of X480 effects
X480 is considered a dual PI3K/mTOR inhibitor. We confirmed this using cell based ELISA in PC3, DU145, and 22rv1 cells used as models for this analysis with IC50 ranging between 250 (PC3) and 750 nM (22rv1) for p-Akt (Ser473, Figure 3(a)), 400 (PC3) and 600 nM (DU145) for p-Akt (Thr308, Figure 3(b)), and 100 (PC3) and 250 nM (DU145) for p-mTOR (Ser 2448, Figure 3(c)). Next, we assessed the antiproliferative activity of X480 on our PCa cell cohort exposed to increasing concentrations of the drugs for different times and then analyzed by direct cell count of viable cell MTS assay. Here we demonstrated that X480 showed antiproliferative effects with IC50 values ranging between 0.4 and 3.5 µM (Figure 3(d)).

Enzymatic inhibition of X480 on PI3K/mTOR activity. The PC3, 22rv1, and DU145 cells were plated in 96-multiwell plates. Cultures were left to adhere and then the complete FBS medium was changed. Growing doses of X480 were added. Cultures were left for 30 min and then treated using cell-based ELISA following protocol from Abcam (see Methods). (a) Cell-based ELISA for p-Akt (Ser473). (b) Cell-based ELISA for p-Akt (Thr308). (c) Cell-based ELISA for p-mTOR (Ser2448). (d) X480 shows antiproliferative effects on PCa cells to various extents depending to levels of enzymatic activities. IC50 values were calculated for X480 administration in our PCa cell cohort. Data are representative of an individual experiment performed in quintuplicate.
PK analysis
Plasma concentration of X480 was measured in nude mice at different time points after multiple p.o. administrations and data were graphically presented in Figure 4. X480 was fast absorbed in nude mouse as indicated by Tmax value, the T1/2 ranged from 1.5 to 3.3 h, and plasma elimination half-life was moderated. Further in vivo studies on tissue distribution/accumulation are in progress.
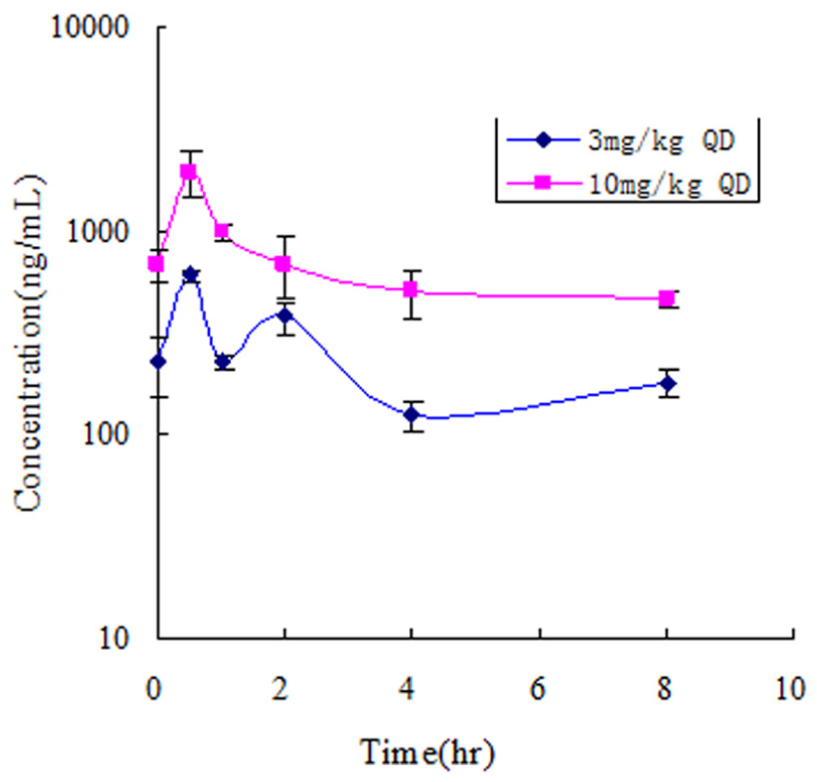
PK analysis. The mean plasma concentration–time curve of X480 after multiple p.o. administration (two dose levels daily for 11 days) in Balb/c nude mice.
X480 determines Tumor Growth delay in PC3 and 22rv1 Xenograft models
PC3 and 22rv1 cells were xenografted in male CD1 nude mice and tumor-bearing mice were treated every day with 3 mg/kg X480. Tumor growth was monitored for 35 days, then all animals were sacrificed, due to the over-growth of untreated controls, and weighed (Figure 5(a) and (c) for PC3 and 22rv1 xenografts, respectively). In order to reduce the error due to differences in tumor volume at the start of drug administration, we analyzed the time to progression (TTP) of single tumors and considered the percentage of tumor with progression by Kaplan–Meier curves (Figure 5(b) and (d)) as previously described.16,18 Results indicated that X480 was more effective in PC3 tumors (PTEN negative) when compared to 22rv1 tumors (PTEN positive). Microscopic appearance of PC3 tumors (Figure 5(e)) stained with Martius yellow-brilliant crystal scarlet blue technique or trichromic stain revealed the presence of numerous phagocytes (neutrophil and macrophages) dispersed in collagen I or fibrin deposits suggesting a previous colliquative necrosis (yellow/orange staining). Immunohistochemical evaluation indicated that X480 inhibited completely Akt signaling also in vivo (Figure 5(f)). The levels of rictor and raptor, instead, were only slightly reduced by treatments.
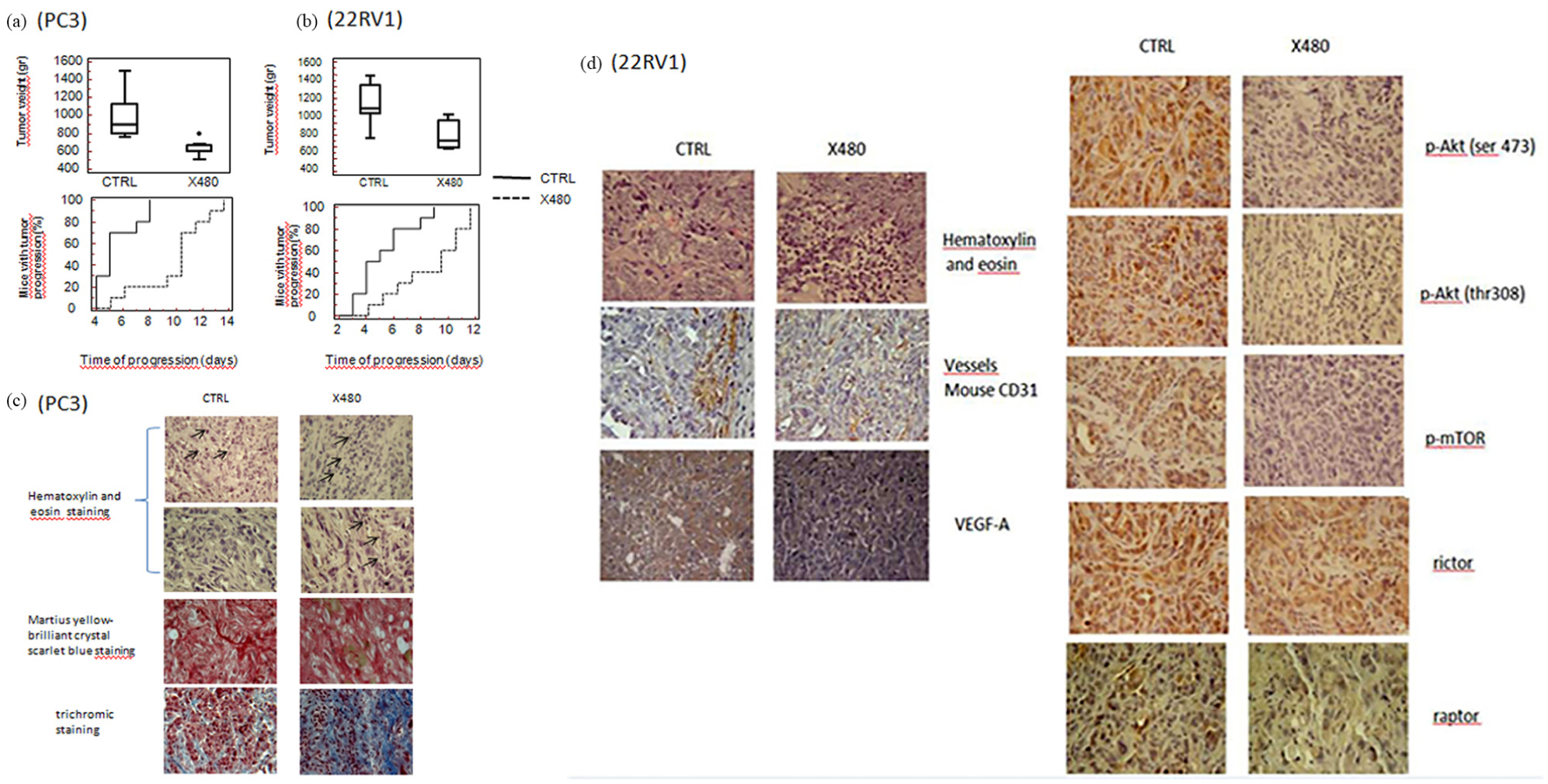
X480 results in tumor growth delay in PC3 and 22rv1 xenograft models: (a) tumor weight differences and evaluation of time to progression and Kaplan–Meier analyses in PC3 xenografts untreated or treated with X480 (3 mg/kg every day); (b) tumor weight differences and evaluation of time to progression and Kaplan–Meier analyses in PC3 xenografts untreated or treated with X480 (3 mg/kg every day); (c) Martius yellow-brilliant crystal scarlet blue technique and trichromic staining with staining for collagen I and fibrin deposits observed in PC3 xenografts. The presence of numerous lymphoid cells (black arrows) dispensed in the tumor cell nests and in collagen I deposits (see throchromic staining) suggests that a previous colliquative necrosis (yellow/orange staining in Martius yellow-brilliant crystal scarlet blue method) was present; (d) immunohistochemical evaluations of p-Akt ser473, p-Akt Thr408, p-mTOR, rictor, and raptor in 22rv1 xenografts harvested from untreated or X480-treated mice. Magnification 200×.
X480 reduces the incidence of bone lesions and increases overall survival of mice with bone metastases
Mice were treated for 35 days with X480 (3 mg/kg every day) and underwent X-ray evaluation at 10, 35, 42, and 50 days after tumor intracardiac cell injection. Experiment was stopped 80 days after tumor cell injection as indicated in Figure 6(a). Intracardiac injection of different PCa cell lines generates bone metastases with high frequency and the prevalent metastatic site is the hindlimb.48,49 For this reason, we analyzed the presence of metastases in femurs and tibias. In Figure 6(b), we show representative images of tumor lesions. In control mice, osteolytic bone metastases appeared at day 35 post inoculation (5/10, 50%) and incidence progressively increased to 100%, at day 80 (Figure 6(c)). The administration of X480 reduced the percentage of X-ray-positive hindlimbs showing 20% of positivity at day 35 (2/10, 20%) with the highest incidence, reached at day 80, of 60% (6/10). These data suggest that X480 increased the time necessary to have a visible bone lesion (incidence) and Kaplan–Meier curves (Figure 6(c)) showed a hazard ratio (HR) value of 6.84 (P = 0.0052) versus untreated animals (Figure 6(d)). Osteosclerotic areas were observed in some bone lesions, especially in X480-treated animals suggesting a stimulation of osteoblast function and/or a reduced osteoclast function. Then we measured lytic areas by ImageJ software and then observed that bone lysis was significantly higher in control mice when compared with X480-treated animals (Figure 6(e)). Similarly, the serum levels of mTRAP (expressed by osteoclasts, macrophages, and dendritic cells) and CTX, markers of bone reabsorption and indirect measures of osteolysis/osteoclast activity, were significantly reduced after treatment with X480 (Figure 6(f) and (g)) when compared to controls. Post-mortem examination of visceral organs showed the presence of lymph node and lung metastases both in controls and treated animals (Figure 6(h)). Although the number and size of lung metastases were reduced by treatment, the percentage of animals with visceral metastases was not statistically different suggesting that PI3K/mTOR pathways could play a major role in bone dissemination. Kaplan–Meier curves (Figure 6(i)) demonstrated that the lethality of PC3 cells inoculated in mice was delayed after X480 administration with an HR of 4.14 (P = 0.0052) versus untreated animals.
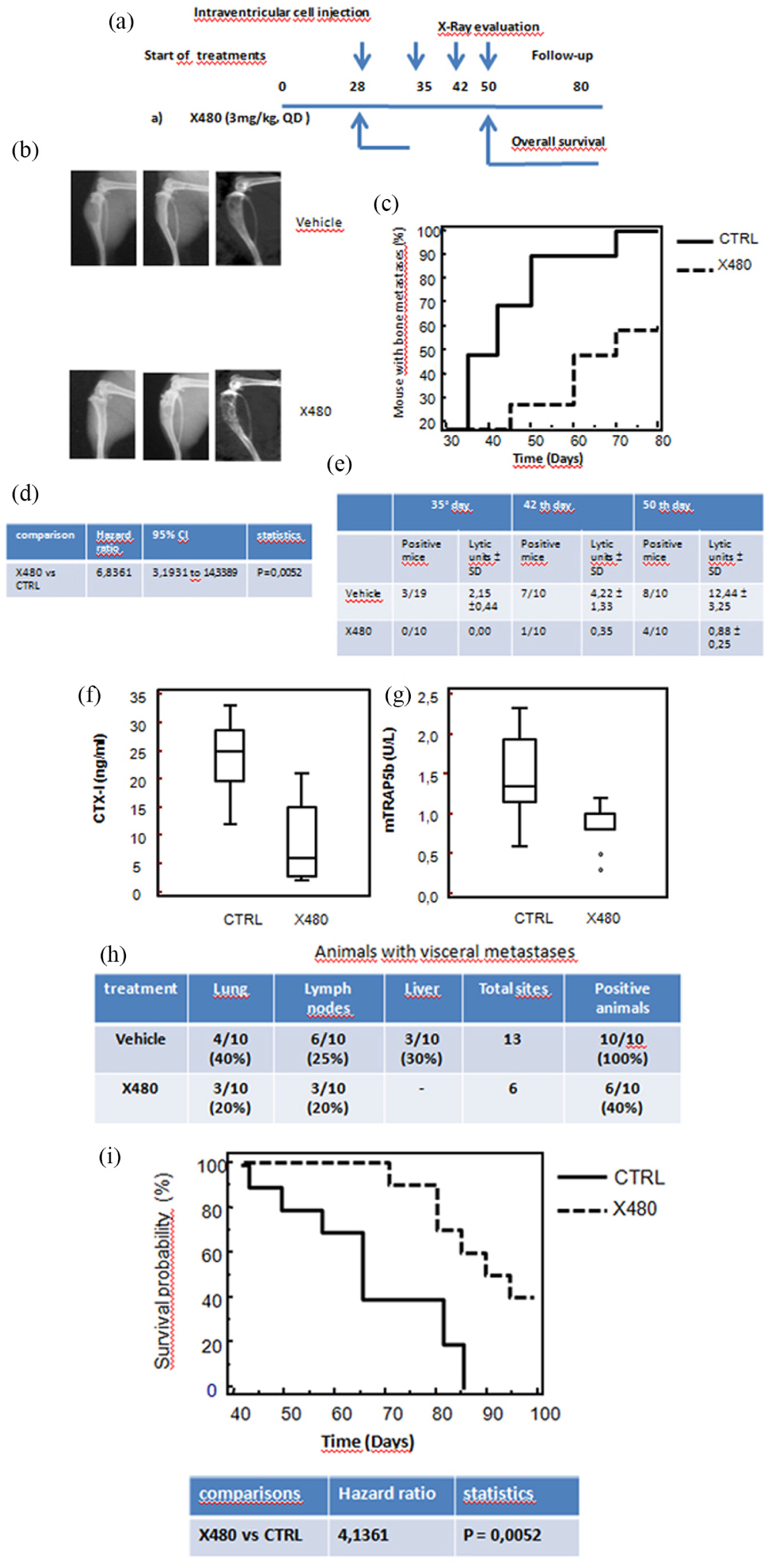
X480 reduces the incidence and size of bone lesions. (a) Scheme of treatments that the mice were treated with for 35 days with X480 (3 mg/kg every day) and underwent X-ray evaluation at 10, 35, 42, and 50 days after tumor cell injection. Experiments were stopped at 80 days when all living animals were euthanized. (b) Representative images of tumor lesions as they appear at Faxitron. (c) Evaluation of bone metastasis incidence (bone marrow disease-free survival (BMDFS)) by Kaplan–Meier analysis. (d) Statistical analysis with hazard ratios calculated for BMDFS. (e) Areas of osteosclerotic areas were quantified on X-rays by measuring lytic areas with ImageJ software. Table of lytic units on determination evaluated at 35, 42, and 50 days after the start of treatments. (f) Serum levels of tartrate-resistant acid phosphatase (TRAP; expressed by activated osteoclasts, macrophages, and dendritic cells). (g) Serum cross laps (a new assay for measuring carboxy-terminal collagen cross links, CTX). X480 reduces visceral metastases and increases overall survival of mice with bone metastases. (h) Quantification of visceral metastases. Post-mortem examination of visceral organs showed the presence of lymph node, liver, and lung metastases in control and treated animals. (i) Overall survival evaluation by analysis of Kaplan–Meier curves with statistical analysis with hazard ratios calculated for OS.
X480 affects tumor growth in bone microenvironment
In order to verify whether the effects of X480 on the reduction of bone metastases incidence were influenced by the reduction of tumor growth rate within bone microenvironment (local growth), PC3 cells were injected directly in the tibia of mice (orthotopic intra-osseous model). This model allowed us to inject a substantially higher number of tumor cells into the bone marrow with respect to intracardiac tumor model and could mimic the high risk of clinical relevant bone metastases. Previously, we demonstrated that 3–5 days after intratibial tumor cell injection, tumor-bearing mice developed inevident (by bioluminescence or X-ray analyses) tumor bone foci. 50 Therefore, we decided to start treatments 5 days after cell inoculation. Skeletal lesions were analyzed by X-ray and μCT and graded as described by Yang et al. 47 as follows: score 0 (absence of tumor growth after X-ray and μCT analyses), score 1 (osteolytic lesions visible only by μCT analyses and a diameter < 5 mm2), score 2 (lesions visible only by μCT and a diameter > 5 mm2), score 3 (lesions visible by X-ray without cortical impairment), score 4 (X-ray visible lesions with cortical impairment with/without fractures), and score 5 (extended osteolysis with extratibial growth). Experiments in Figure 7(a) show representative radiological appearance of bone lesions arranged according to Yang score compared to digitalized µCT images. At 15 days after tumor injection within tibiae, a rate of 90% in the radiographically evident bone lesions was observed. We observed that X480-treated mice showed smaller lytic lesions. Seven out of 10 and 5 out of 10 metastases in the and X480 group had a lytic score ≤ 2, while only 1 out of 10 metastases in untreated animals graded with the same score (Figure 7(b)). Significant differences were found between the control and X480-treated groups (P = 0.06617). The serum analyses indicated decreased levels of osteolytic markers (CTX-I and TRAP5b; Figure 7(c) and (d)) at day 15, which correlated with a positive response to systemic anticancer treatments in terms of reduced osteolysis. As previously demonstrated, 50 untreated mice with skeletal metastases show a time-dependent significant reduction of body weight. Here, we demonstrated that X480 resulted in a not significant time-dependent reduction in body weight loss as reported in Figure 6(e). Next we analyzed the rate of cachexia in untreated and treated mice. When Cochran’s Q test was performed, the rate of cachexia in the untreated mice significantly increased with respect to the baseline (Figure 7(f)).

X480 affects tumor growth in bone microenvironment after intratibial injection. (a) Representative radiological appearance of bone lesions arranged according to Yang score compared to digitalized µCT images. (b) Yang scores for bone metastases. (c) Serum analyses of CTX-I and (d) TRAP5b at day 15, which correlated with a positive response to systemic anticancer treatments in terms of reduced osteolysis. (e) Effects of X480 administration on body weight loss of treated or untreated animals. (f) Statistical analyses by Cochran’s Q test for the rate of cachexia in the untreated and X480-treated mice. These data indicate that the induction of cachexia was significantly dependent on the size of bone metastases that may inhibit the capacity of animals to eat and drink (Cochrane’s Q value = 6.5; P = 0.039), whereas this appearance was not evident in X480 (Cochrane’s Q value = 2.0; P = 0.368).
Reduction of osteoclastogenesis and increased proliferation and activity of osteoblasts
To demonstrate an effect of X480 on osteoblasts, we used two models of murine osteoblast-like cells such as the culture of bone marrow stromal cells and of mouse calvaria. Cells were treated with conditioned media from PC3 cells and non-toxic concentrations of X480 (0.5µM). In these conditions, osteoblastic differentiation was more evident in bone marrow osteoblasts than in osteoblasts from the calvaria where X480 seemed to have inhibitory effects (Figure 8(a)). The analysis of cell proliferation, however, demonstrated that the number of cells and the enzymatic activity (ALP; Figure 8(b)–(d)) were significantly lower when compared with the growth and differentiation of bone stromal cell cultures. In addition, considering that the microenvironment which was found by PC3 cells was produced by bone marrow stromal cells, we might conclude that X480 might increase osteoblastic differentiation. So murine osteoclast precursor cells RAW 264.7 were cultured with the conditioned medium (CM) collected from bone-derived PC3 cells and TRAP positive multinucleated cells were counted. Within 24 h of culturing, RAW 264.7 cells started to differentiate into multinuclear cells and at 96 h the PC3 CM induced TRAP-positive multinuclear cell formation (Figure 8(f)). We found that X480 inhibited the osteoclast differentiation both when RANKL was used as the osteoclast differentiation factor and when PC3 CM was used. Interestingly, the CM harvested from PC3 cells treated with X480 was able to reduce osteoclastogenesis suggesting also the presence of an indirect paracrine effect on PCa cells.

(a) Evaluation of osteogenic potential of PC3 CM treated or not with X480. Osteogenic potential was verified using bone marrow stromal cell cultures cultured with osteogenic medium (CTRL +) with or without 0.5µM X480. After treatments and at the times indicated in material and methods, the cultures were stained using Alizarin Red S staining kit and absorbance was measured at 492 nm as indicated by the manufacturer. Osteoblasts derived from the calvaria were also used as more differentiated models. (b) Effects of 0.5µM X480 on the growth of osteoblast from the calvaria (optical density (OD): 450 nm). (c) Counts of ALP-positive cells for microscopic field normalized on total cell number (percentage of ALP + cells). (d) Quantification of mRNA for osteonectin versus glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by semi quantitative RT-PCR and (e) quantification of mRNA for collagen 1a2 versus GAPDH by semi quantitative RT-PCR. Both markers are considered as osteoblast markers. (f, g) Evaluation of osteolytic potential: activation of osteoclasts from RAW 274 cells.
Discussion
Bone metastasis is a frequent event in patients with mCRPCa and prevention would likely increase both survival and quality of life of these patients. Overexpression and increased activity of MCP-1, SDF1α, and IL8 have been strongly implicated in the progression of PCa.51–55 In addition, it has been demonstrated that increased TNFα may sustain a non-permissive or “hostile” stromal microenvironment which actively prevents tumor engraftment in vivo. 50 Altogether, these signaling pathways increase the aberrant expression of PI3K/Akt/mTOR-related molecules which have been considered to facilitate cell growth, invasion, adhesion to bone matrix, bone metastasis, and angiogenesis as well as increase pharmacological and radiation therapy resistance. Our immunohistochemical and molecular data seem to confirm the evidence that the expression levels and phosphorylation status of Akt and its downstream effectors are preferentially increased in PCa cells derived from bone metastatic lesions as well as in bone metastatic human tissues. Our in vitro data suggest also that the interaction of tumor cells with some components of bone marrow microenvironment may greatly increase the mTOR activity. So, we argue that, when tumor cells reach the bone and interact with bone marrow microenvironment, a sustained increase in SDF1-α and TGF-β1 happens and this, in turn, favors the increase of tumor CXCR4. Next a change in the “inflammatory status” increases the insurgence of a pro-tumor microenvironment and establishment of favorable highly hypoxic endosteal niche. In the pre-metastatic niche, the bone is lined with osteoblasts which secrete homing factors such as SDF1α/CXCL12. 56 The endosteal hematopoietic stem cell (HSC) niche also comprises myeloid mononucleated cells (MDSCs) which are recruited into the pre-metastatic niche, 57 where they can influence the differentiation of osteoblasts or osteoclasts. 49 Also the intratibial model can suggest an interfering role of X480 in the metastatic homing; in fact, it is well known the presence of an endosteal niche that allows the growth of PCa cells. In this study, we demonstrated the dual PI3K/mTOR inhibitor X480 reduced PCa growth in primary and bone metastatic sites altering the metastatic niche and reducing the activation of osteoclasts with decreased intratibial tumors. Tumor cells directly affect the immune system and induce an immunosuppressive environment, for example, by recruiting and activating MDSCs and shifting the phenotype of tumor-infiltrating macrophages, which increases the possibility for tumor cells to grow. It has been demonstrated that chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs the life of immune-deficient mice. 58 The pro-inflammatory tumor microenvironment is generally considered to be anti-tumorigenic, stimulating the immune system to kill the tumor cells, whereas immunosuppressive functions help the tumor to evade the immune response and to grow and metastasize. Interactions between osteoblasts and cancer cells are believed to be important for the establishment and development of PCa bone metastases. Osteoblasts have been shown to induce a more aggressive and osteoblast-like phenotype in PCa cells through soluble factors and cancer cells, in turn, influence osteoblast proliferation and differentiation. 5 In this study, X480 increased osteoblast differentiation in vitro and reduced osteoclast viability and differentiation.
Conclusion
A growing body of evidence has demonstrated that the PI3K/Akt/mTOR pathway plays an important role in cancer proliferation, dissemination, and invasion and our results seem to confirm its role in primary growth and bone metastasis formation. Our report is the first demonstrating that the blockade of PI3K/TORC2 activity by a dual PI3K/mTOR inhibitor results in significant prolongation of survival in animal models of PCa bone metastases. Our data support a biological rationale for the use of this inhibition strategy in men with clinically evident bone lesions as well as in men at high risk of developing bone metastases.
Footnotes
Acknowledgements
We thank George Liu and Xiaobao Xu at the Sundia MediTech Facility for the pharmacokinetic analyses (Study Project: SMC-XC-040111, DMPK, sponsored by Xcovery, LLC, West Palm Beach, FL). Giovanni Luca Gravina and Claudio Festuccia contributed equally. Chris Liang is an employee of Xcovery LLC, West Palm Beach, FL and Xcovery LLC has partially contributed to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partially supported by Xcovery, LLC, West Palm Beach, FL and by the ALCLI “Giorgio e Silvia” Non-Profit Association.