
Abstract
The Src homology-2 domain protein B is an adaptor protein operating downstream of tyrosine kinases. The Shb gene knockout has been found to accelerate p210 Breakpoint cluster region-cAbl oncogene 1 tyrosine kinase-induced leukemia. In human myeloid leukemia were tumors with high Src homology-2 domain protein B mRNA content, tumors were, however, associated with decreased latency and myeloid leukemia exhibiting immune cell characteristics. Thus, the aim of this study was to investigate the effects of Shb knockout on the development of leukemia in three additional models, that is, colony stimulating factor 3 receptor-T618I–induced neutrophilic leukemia, p190 Breakpoint cluster region-cAbl oncogene 1 tyrosine kinase-induced B-cell leukemia, and G12D-Kras-induced T-cell leukemia/thymic lymphoma. Wild-type or Shb knockout bone marrow cells expressing the oncogenes were transplanted to bone marrow–deficient recipients. Organs from moribund mice were collected and further analyzed. Shb knockout increased the development of CSF3RT618I-induced leukemia and increased the white blood cell count at the time of death. In the p190 Breakpoint cluster region-cAbl oncogene 1 tyrosine kinase B-cell model, Shb knockout reduced white blood cell counts without affecting latency, whereas in the G12D-Kras T-cell model, thymus size was increased without major effects on latency, suggesting that Shb knockout accelerates the development thymic lymphoma. Cytokine secretion plays a role in the progression of leukemia, and consequently Shb knockout bone marrows exhibited lower expression of granulocyte colony stimulating factor and interleukin 6 in the neutrophilic model and interleukin 7 and chemokine C-X-C motif ligand 12 (C-X-C motif chemokine 12) in the B-cell model. It is concluded that in experimental mouse models, the absence of the Shb gene exacerbates the disease in myeloid leukemia, whereas it alters the disease characteristics without affecting latency in B- and T-cell leukemia. The results suggest a role of Shb in modulating the disease characteristics depending on the oncogenic insult operating on hematopoietic cells. These findings help explain the outcome of human disease in relation to Src homology-2 domain protein B mRNA content.
Keywords
Introduction
Mutation of kinase and growth factor signaling pathways is common in hematological malignancies, and certain genetic abnormalities may give rise to different kinds of leukemia (e.g. Breakpoint cluster region-cAbl oncogene 1 tyrosine kinase (BCR–ABL), NRAS, and KRAS mutations that are observed in both lymphoid and myeloid malignancies). The capacity of one oncogene to drive differential disease phenotypes suggests that certain elements may be common between different forms of leukemia and that disease will be directed toward different paths due to superimposed additional genetic/environmental aberrations. Src homology-2 domain protein B (SHB) is a pleiotropic adaptor protein that transduces signals from receptor tyrosine kinases to intracellular intermediates regulating numerous responses.1–4 We have previously investigated the relevance of Shb deficiency in p210 BCR–ABL myeloid leukemia and observed that the absence of Shb in this model results in the accelerated development of disease, due to increased focal adhesion kinase (FAK) activity, increased cytokine production, and activation of signal transducer and activator of transcription 3 (STAT3). 5 In human acute myeloid leukemia (AML), high expression of the SHB gene was associated with shorter survival. 6 The “SHB high” AML patients have a tumor gene expression profile that displays immunological features. For this reason, it can be hypothesized that the Shb gene plays a role in bridging phenotypic traits between different forms of leukemia. Consequently, it would be of interest to investigate the role of Shb in additional types of leukemia that have distinct phenotypic and genetic characteristics to determine how Shb influences disease outcome in multiple settings. SHB may regulate the activities of FAK, c-Abl oncogene 1 tyrosine kinase (ABL), and extracellular signal-regulated kinase (ERK, also named mitogen-activated protein kinase (MAPK)) in response to receptor tyrosine kinase activation, providing ample possibilities for SHB to modulate the phenotypic characteristics of leukemic cells.2,7 It has been shown previously that the absence of Shb leads to alterations in T-cell receptor (TCR) signaling8–10 and that Shb also is required for normal hematopoietic stem cell (HSC) proliferation. 9 The SHB protein has thus been demonstrated to specifically regulate important functions for hematopoietic cells.
The BCR–ABL fusion gene is one important cause of leukemia and it results from a genetic aberration called the Philadelphia chromosome. This chromosomal translocation of t(9;22)(q34;q11) may generate three different BCR–ABL fusion proteins (p190, p210, and p230) that have different characteristics.11,12 The p190 isoform is typically associated with B-cell acute lymphoblastic leukemia (B-ALL), the p210 most commonly with chronic myeloid leukemia (CML) but occasionally with B-ALL, and the rare p230 isoform is associated with a CML-variant that usually exhibits neutrophilia and/or thrombocytosis. One noticeable consequence of Shb deficiency in the p210 BCR–ABL model was the expansion of mature neutrophilic granulocytes. This suggests a possible connection between Shb and BCR–ABL-dependent neutrophilia. Chronic neutrophilic leukemia (CNL) was officially classified as a tumor in 2001 by the World Health Organization (WHO). 13 Mutations in the colony stimulating factor 3 receptor (CSF3R) are commonly observed in BCR–ABL-negative CNL,14–16 and CSF3R mutation has been included in the 2016 WHO revision of diagnostic criteria for CNL. 17 Studies have identified a proximal point mutation CSF3RT618I in a noticeable percentage of patients suffering from CNL and atypical (BCR–ABL-negative) chronic myeloid leukemia (aCML). 16 The CSF3RT618I product has a ligand-independent function, resulting in constitutively activated CSF3R and, consequently, massive production of neutrophils due to differentiation and maturation of granulocyte progenitors.16,18 CSF3R has been shown to activate the Janus kinase (JAK)/STAT signaling pathways 19 with activation of STAT3 as a prominent feature. The neutrophilia observed after p210 BCR–ABL-induced leukemia in the absence of Shb stimulated us to investigate Shb-dependent disease characteristics also in CSF3RT618I-induced CNL, whereas the immune cell phenotype observed in human AML patients expressing “high SHB” motivated an investigation of Shb deficiency in p190 BCR–ABL-induced B-cell leukemia.
Kras is one of the most common mutations in human cancers, including hematopoietic malignancies. Oncogenic Kras is primarily found in human myeloid and T-cell leukemias. 20 In addition, the elevated level of rat sarcoma (RAS) signaling has been observed in AML and T-ALL.21,22 Oncogenic mutations in the Ras genes result in constitutive activation of RAS proteins. 23 RAS is an essential factor for the signaling pathways underlying cell proliferation, differentiation, and survival by triggering the v-raf1-murine leukemia viral oncogene homolog (RAF)/mitogen-activated protein kinase kinase (MEK)/MAPK pathway. 23 In an experimental murine model for T-ALL or T-cell thymic lymphoma, bone marrow cells from mice carrying active KrasG12D on the Kras2LSL allele are transplanted to wild-type recipients that subsequently develop disease.24,25 The fact that SHB operates downstream of the TCR stimulated us to investigate the effects of Shb deficiency in the KrasG12D T-cell model.
Based on the findings observed in p210 BCR–ABL-induced leukemia in mice and in relation to SHB mRNA contents in human AML, we considered it prudent to investigate Shb in additional mouse models. The data presented show the effects of Shb deficiency in three experimental mouse models of leukemia: CSF3RT618I CNL, p190 BCR–ABL B-ALL, and oncogenic G12D-Kras T-ALL/thymic lymphoma. In all three models, the disease characteristics were altered, albeit in a fashion unique for each model. The data support the notion that the expression of the Shb gene plays a role in defining human disease phenotypes, possibly by bridging myeloid and lymphocytic features in leukemia.
Methods
Mice
Shb wild-type or knockout mice on the Balb/c background were used for the induction of p190 BCR–ABL- and CSR3RT618I-induced leukemia. KrasLSL and Mx1-Cre mixed background mice were kindly provided by Professor Martin Bergö at Sahlgrenska Cancer Center, University of Gothenburg, and crossed with Shb-null mice to generate KrasMxCre-Shb knockout triple-transgenic mice. The bone marrow cells isolated from these mice after the induction of MxCre will be named wild-type or Shb knockout KrasG12D throughout the text. The experiments were ethically approved by the local animal ethics committee at Uppsala University (approval number C22/14).
Leukemia induction
For CSF3RT618I-induced neutrophilic leukemia, the MIGR1 retroviral vector was used for producing CSF3RT618I-GFP retroviruses in 293T/17 human embryonic kidney (HEK) cells.26,27 Balb/c wild-type or Shb knockout mice at 8–10 weeks of age were injected intraperitoneally with 5-fluorouracil (5-FU; Sigma-Aldrich, St Louis, MO) at a dose of 150 mg/kg body weight 6 days prior to bone marrow isolation, in order to enrich the HSC pool. Isolated donor bone marrow was then cultured in RPMI 1640 (Sigma-Aldrich), supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich), 2 mM L-glutamine, streptomycin (0.1 mg/mL), and penicillin (100 U/mL) (All from Gibco), interleukin 3 (IL-3; 10 ng/mL), stem cell factor (SCF; 10 ng/mL), and interleukin 6 (IL-6; 10 ng/mL) (all cytokines from PeproTech). The cells were subsequently spin-infected twice over the following 48 h. Determination of infection efficiency was assessed by fluorescence-activated cell sorting (FACS) analysis of green fluorescent protein (GFP) expression on a FACSCalibur (BD Bioscience). Wild-type and Shb knockout bone marrow were similarly infected (16% ± 5.1% in wild type; 12.7% ± 3.8% in Shb knockout). Wild-type recipient mice were lethally irradiated with two doses of 4.5 Gy in a Nordion Gammacell 40 Exactor 137Cs irradiator (MDS Nordion), with at least 2-h interval. The recipients were subsequently injected retroorbitally with a dose of 0.5–1 × 106 cells with the same number of wild-type and Shb knockout cells injected in each experiment, that is, transfection/transplantation event.
For p190 BCR–ABL-induced B-cell leukemia, retroviruses expressing p190 BCR–ABL-GFP were produced using pMIG-p190 BCR/ABL vector transfected to 293T/17 HEK cells. 26 Bone marrow cells were isolated from 8- to 10-week-old Balb/c wild-type and Shb knockout donor mice and stimulated in RPMI 1640 cultured in 10% FCS, 2 mM L-glutamine, streptomycin (0.1 mg/mL), penicillin (100 U/mL), IL-3 (10 ng/mL), SCF (10 ng/mL), IL-6 (10 ng/mL), and interleukin 7 (IL-7; 10 ng/mL) for 24 h. The cells were then spin-infected twice over the following 48 h. The infected cells were kept in culture with the aforementioned sufficient cytokines for 5 or more days and the media were changed every 2–3 days. Infection efficiency was determined prior to transplantation by flow cytometric analysis of GFP expression on a FACSCalibur and no differences were found between wild-type and Shb knockout bone marrows (45.25% ± 3.3% in wild type; 49.25 ± 5% in Shb knockout). Notably, almost all (>90%) of the cells expressed mature B-lymphocyte markers (CD19, B220) prior to transplantation. Recipient wild-type mice were sublethally irradiated with 5.5 Gy, 24 h prior to the transplantation. The animals were retroorbitally injected with a dose of 1.25–4 × 106 cells. The equal numbers of wild-type and Shb knockout cells were given per recipient mouse in each experiment, that is, transfection/transplantation event.
For G12D-Kras T-cell leukemia/thymic lymphoma, KrasMxCre wild-type and KrasMxCre-Shb knockout mice at 4 weeks of age were injected intraperitoneally with polyinosinic–polycytidylic acid (Poly I:C; Sigma-Aldrich) for four consecutive days, at a dose of 400 µg (200 µl) per day to induce floxing of KrasLSL leading to the expression of KrasG12D from the endogenous Kras locus. 28 Bone marrows were isolated 4–5 weeks post injection and frozen in 10% dimethyl sulfoxide (DMSO) at −135°C. Wild-type mice were lethally irradiated with two doses of 4.5 Gy with at least 2-h gap in between, followed by retroorbital injection of the thawed bone marrow cells having the KrasG12D on wild-type or Shb knockout genotypes at a dose of 2 × 106 cells per mouse.
Mice having received leukemic bone marrow cells were checked daily for any signs of disease, starting 6 days after transplantation. Body weight was regularly measured and blood smears were collected at 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, and 120 days post transplantation. The mice were monitored for signs of disease such as a maximal weight loss of 20% relative to the weight at injection of the bone marrow cells, lethargy, unkempt fur, infections, expanded abdomen, uneven respiration, and abnormalities in locomotion such as a limp. Moribund mice were immediately sacrificed and analyzed. The experimental endpoint was set to 19 weeks after injection of the bone marrow cells. Mice that showed no signs of disease at this point were euthanized and their tissues processed for assays described below.
Blood was collected immediately prior to sacrifice for preparing blood smears, counting blood cells, and for further experiments. The lungs were monitored for possible hemorrhage. Spleens, thymi, and lymph nodes as well as iliac bones, femurs, and tibia were dissected and the cells of these organs were isolated. Single-cell suspensions of the organs were fixed in 4% paraformaldehyde to enable FACS analysis at a later time point.
Whole blood profile
Peripheral blood from moribund mice was collected in Microtainer K2E tubes (BD). The blood was subsequently analyzed using a Sysmex XP-300™ Automated Hematology Analyzer (Sysmex, USA). Peripheral blood smears were stained with May-Grünwald Giemsa (Merck Millipore) based on the manufacturer’s protocol.
RNA isolation and real-time reverse transcription polymerase chain reaction
Bone marrow c-Kit+ cells were isolated using magnetic separation with anti-c-Kit-labeled magnetic microbeads (Miltenyi Biotec), in order to enrich bone marrow for hematopoietic stem and progenitor cells. The c-Kit+ cells were counted and RNA was subsequently extracted using RNeasy mini kit (Qiagen). Analysis of gene expression was conducted through real-time reverse transcription polymerase chain reaction (RT-PCR) using the QuantiTect™ SYBR® Green RT-PCR kit (Qiagen) according to the following conditions: reverse transcription at 50°C for 20 min, inactivation at 95°C for 15 min, 50 cycles of denaturation at 94°C for 15 s, annealing at 60°C for 25 s, and extension at 72°C for 15 s. The polymerase chain reactions (PCRs) were all run on a LightCycler™ real-time PCR machine (Roche Diagnostics). The cycle threshold (CT) values were estimated with the LightCycler Software v4.1 and the transcript levels were normalized by subtracting the corresponding β-actin values. The expression differences were then presented as 2–ΔKOCt–WTCt.
FACS analysis
In order to identify B-lymphocyte, T-lymphocyte, and myeloid cell population, paraformaldehyde-fixed peripheral blood cells as well as bone marrow, spleen, thymus, and lymph node single cells were stained with antibodies recognizing lineage markers, such as B220 (eBioscience) and CD19 (BioLegend) for B cells, biotin rat anti-mouse CD3, CD4, and CD8 for T cells (BD Pharmingen), or Gr-1 (Life technologies) and CD11b (Invitrogen) for myeloid cells. The cells were thereafter incubated with biotin goat anti-rat IgG secondary antibody (Invitrogen) when staining for the B-cell and myeloid markers, followed by incubation with PerCP/Cy5.5 streptavidin (BioLegend) in all stains. HSC populations were identified by excluding the cells expressing lineage markers while simultaneously staining for CD150-PE-Cy7 (BioLegend), c-Kit-APC eFluor 780, Flk2-PE, CD41-PE, CD48-PE (eBioscience), and Hoechst 33342 (Sigma-Aldrich). To identify T-lymphocyte development in thymus, quadruple staining of single thymocyte suspension was done by CD25-PE, CD44-PE/Cy5 (eBioscience), CD4-FITC (BioLegend), and biotin CD8 (BD Pharmingen), followed by incubation with Alexa Fluor 680 streptavidin (Life Technologies).
All flow cytometric experiments were analyzed with an LSRFortessa (BD Bioscience) and the data were analyzed with FlowJo (TreeStar).
Immunoblotting
The c-Kit+ cells, enriched from the bone marrow, were lysed in SDS sample buffer (3M Tris, pH 8.8, 2% SDS, 10% glycerol, 0.006% bromophenol blue, 2% β-mercaptoethanol). Separation of the samples was done by SDS-PAGE and then transferred to a Hybond-P membrane (GE Healthcare). The membrane was blocked at 4°C in 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS), including 0.5% Tween 20 as well as 0.1% azide, followed by probing for phospho-FAK (p397-FAK, 44624G; Invitrogen), phospho-ERK (pERK, 9101S; Cell Signaling Technology), phospho-protein kinase B (pAKT and total v-akt murine thymoma viral oncogene homolog (AKT), 09E; Cell Signaling Technology), ERK (SC-94; Santa Cruz), and FAK (A-17; Santa Cruz). Bands were subjected to densitometry and background signal was determined by placing an equally large rectangle as that used for the assessment of the positive band’s signal over an area without bands in a close proximity to the relevant band. Background was then subtracted before the calculation of pFAK, pERK, and pAKT relative to total ERK, FAK, and AKT.
Statistical analysis
All values are presented as mean ± standard error of mean (SEM). Kaplan–Meier survival analysis was performed and survival differences between wild-type and Shb knockout transplanted mice were estimated with the log-rank test, assuming significance at p < 0.05. Unpaired 2-tailed Student’s t test was used for comparison between two groups. Statistical significance was set at p < 0.05.
Results
The development of CNL induced by CSF3RT618I is accelerated by the absence of Shb
Studies have demonstrated the presence of a membrane proximal point mutation, T618I in CSF3R, in many patients exhibiting CNL, a disease that is characterized by a high number of neutrophils in blood. 16 The neutrophilia observed in Shb-deficient p210 BCR–ABL-induced leukemia prompted us to investigate the consequences of CSF3RT618I-expressing hematopoietic bone marrow cells from wild-type and Shb knockout mice that were transplanted to irradiated wild-type recipients. Peripheral blood cell counts from day 15 and onwards revealed no major differences between the genotypes (Supplementary Material Figure 1(a)). A large proportion of leukocytes were morphologically distinct neutrophilic granulocytes (Supplementary Material Figure 1(b); 44% ± 6% wild-type and 42% ± 6% knockout, respectively). A comparison of survival times revealed that the mice receiving Shb-null bone marrows presented with shorter latency (p < 0.01; Figure 1(a)). CNL due to CSF3RT618I is associated with abnormal bone marrow hypercellularity, mostly containing mature granulocytes. The consequence of this hypercellularity is elevated numbers of neutrophils in blood and infiltration of granulocytes into the spleen and liver. 27 Mice receiving Shb knockout leukemic bone marrow displayed larger livers compared to their wild-type equivalents, whereas the spleen size, weight loss, and bone marrow cellularity were unaffected (Figure 1(b) and (c)). When comparing the number of white blood cells (WBCs) at the time of death between wild-type and Shb-null recipients, the latter were found to exhibit significantly increased numbers of leukocytes (Figure 1(d)). The numbers of red blood cells (RBCs) and platelets were not significantly different between wild-type and Shb knockout transplanted mice (Figure 1(e) and (f)).

Absence of Shb and CSF3RT618I CNL. (a) Kaplan–Meier survival curve of recipients of wild-type and Shb-null CSF3RT618I-expressing hematopoietic bone marrow cells. Significant difference was determined by log-rank test. (b) Evaluation of major factors in progressive leukemia, such as weights of the liver and spleen plus weight loss when moribund. (c) Number of bone marrow cells of iliac, tibia, and femur bones of recipient mice at the time of death. (d)–(f) Analysis of blood profile, indicating the numbers of WBCs, RBCs, and platelets. FACS analysis indicating the percentage of lineage defining marker-expressing cells in (g) peripheral blood and (h) bone marrow of transplanted mice with wild-type and Shb knockout CSF3RT618I-expressing bone marrow cells. Results are presented as mean values ± SEM from 12 mice (a–g) and 9 mice (h) of each genotype in 4 and 3 independent experiments, respectively. * denotes p < 0.05 as determined by Student’s t test.
Flow cytometry of peripheral blood revealed a decreased proportion of B cells and a corresponding increase in the percentage of cells staining for myeloid cell markers in Shb-null recipients (Figure 1(g); Supplementary Material Figure 2(a) and (b)). The level of T cells was similar in wild-type and Shb knockout blood (Figure 1(g); Supplementary Material Figure 2(c)). However, the knockout mice had an elevated percentage of lineage-positive cells compared to their wild-type counterparts, suggesting relatively mature WBC populations in peripheral blood. FACS analysis of bone marrow B-cell, myeloid, and T-cell markers revealed no significant differences when comparing wild-type and Shb-deficient CSF3RT618I-expressing mice (Figure 1(h)). It thus appears that the absence of Shb drives the accumulation of increased amounts of relatively mature myeloid cells in blood from CSF3RT618I-expressing bone marrow.
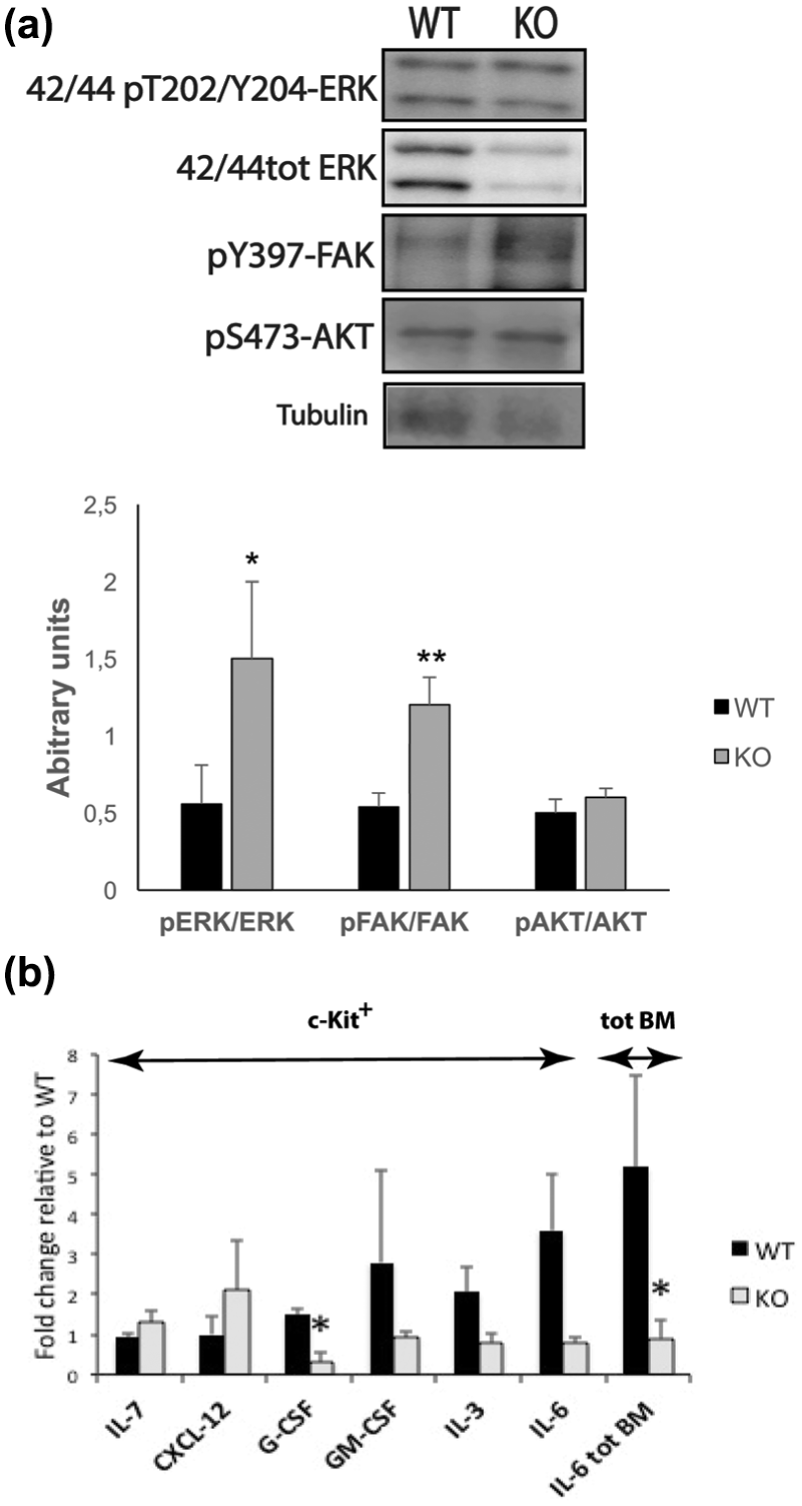
Signal transduction and cytokine expression in the absence of Shb in CSF3RT618I CNL. (a) Western blot analysis of phosphorylated FAK, ERK, and AKT and total ERK in total bone marrow of leukemic mice receiving wild-type and Shb knockout CSF3RT618I-expressing cells. The same two tumor samples are shown. Signal strength was evaluated by densitometric analysis and related to total ERK, total FAK, and total AKT as indicated. Values are presented as relative expression ± SEM, based on five mice of each genotype in two independent experiments. (b) Hematopoietic cytokine expression level in c-Kit-enriched and total bone marrow of CSF3RT618I-induced leukemic recipients. It has been determined by semi-quantitative real-time RT-PCR. The CT values were normalized to β-actin standards and Shb knockout cells were associated to the corresponding wild-type values. The expression differences were then determined as 2–ΔKOCt–WTCt ± SEM to identify fold change in mRNA level. Values are based on c-Kit+ cells from three mice of each genotype in one experiment and the total bone marrow cells of five mice per genotype in two experiments. * and ** denote p < 0.05 and p < 0.01, respectively, as confirmed by Student’s t test.
In p210 BCR–ABL-induced myeloid leukemia, Shb deficiency was found to increase the FAK and STAT3 activities. 5 We thus decided to investigate bone marrow signal transduction in the current setting as well. Shb-deficient bone marrow exhibited increased phosphorylation of FAK and ERK activation sites, whereas no effect on AKT was noted (Figure 2(a)). STAT3 activation/phosphorylation was too low in the present setting to allow accurate detection and thus STAT3 phosphorylation is not shown. It has been demonstrated that cytokines have crucial roles in promoting hematopoietic cell proliferation and inhibiting apoptosis. 29 To explore the expression levels of cytokines that could potentially regulate leukemogenesis, quantitative polymerase chain reaction (qPCR) was performed. The expression levels of cytokines including Il-7, Cxcl-12, Csf2, and Il-6 in c-Kit+ bone marrow cells did not differ between wild-type and Shb knockout transplanted bone marrow, while Csf3 was expressed at a lower level in the absence of Shb (Figure 2(b)). The Il-6 mRNA level tended to be decreased in Shb knockout c-Kit+ bone marrow cells; therefore, the expression level of Il-6 in total bone marrow was examined as well. Consequently, Shb-deficient bone marrow expressed less of Il-6 compared to the wild type implying that more mature cell types were primarily responsible for the changes in IL-6 expression.
Shb deficiency alters disease characteristics of p190 BCR–ABL-induced B-cell leukemia
Considering the fact that Shb deficiency decreases latency in two models of myeloid leukemia, we found it appropriate to investigate disease characteristics in models of B-cell and T-cell leukemia as well. Wild-type and Shb knockout bone marrow cells expressing p190 BCR-ABL were transplanted to induce B-cell leukemia. Upon following the animals post engraftment, no significant difference in latency of wild-type and knockout mice was observed (Figure 3(a)). Mice receiving knockout leukemic bone marrow displayed a larger weight loss at the late stages of disease. In contrast, their liver size was smaller, whereas no difference in the spleen size between wild-type and Shb knockout recipients was observed (Figure 3(b)). The enlarged liver in wild-type leukemic mice might be an explanation for the lower weight loss when moribund. The number of bone marrow cells did not differ between the mice transplanted with transformed wild-type or knockout bone marrow (Figure 3(c)). Analysis of peripheral blood WBCs revealed a decreased number of cells in mice receiving Shb knockout transformed bone marrow (Figure 3(d)), whereas the number of RBCs as well as platelets was unchanged (Figure 3(e) and (f)). In addition, the percentage of morphologically distinct neutrophilic granulocytes in peripheral blood was significantly higher (41% ± 7%) in the absence of Shb expression when compared with that of wild type (21% ± 5%, p < 0.05).

Absence of Shb and p190 BCR-ABL leukemia. (a) Kaplan–Meier survival curve of mice subjected to experimentally induced p190 BCR-ABL leukemia on wild-type or Shb-deficient backgrounds. (b) Evaluation of main disease factors, such as the liver and spleen weights as well as weight loss at the time of death. For weight loss, the mean weight loss of each genotype was determined in each separate experiment. These differences were then compared by paired Student’s t test. Figures (a) to (f) are based on four independent experiments with a total of 13 mice per genotype. (c) Number of bone marrow cells of the iliac, tibia, and femur bones of recipient mice at end stage. (e) and (f) Analysis of blood profile, determining the numbers of WBCs, RBCs, and platelets. FACS analysis showing the percentage of B-cell, myeloid, and lineage+ cells in (g) peripheral blood and (h) bone marrow of transplanted mice with wild-type and Shb knockout p190 BCR-ABL-expressing bone marrow cells. Results are presented as mean values ± SEM from 9–10 mice of each genotype in three independent experiments, respectively. * and ** denote p < 0.05 and p < 0.01, respectively, as confirmed by Student’s t test.
Analysis of in vitro B-cell development after retroviral transduction but prior to transplantation revealed the expression of B220 and CD19 in >85% of the cells, suggesting a population of cells that has the potential to differentiate into B cells. FACS analysis of peripheral blood from moribund mice showed that the percentage of mature B-lymphocytes was significantly higher in the blood of animals without Shb expression compared to their wild-type counterparts (Figure 3(g)). Although the percentage of cells expressing myeloid markers was similar between the two genotypes, the sum of cells expressing these lineage-defining markers was significantly higher in Shb-null bone marrow, suggesting a more mature cell population in the absence of Shb also in this disease model. In the corresponding bone marrows, no differences could be detected (Figure 3(h)). Due to the short latency of p190 BCR-ABL-induced B-cell neoplasia, T-cell differentiation has not yet occurred 30 and thus we were unable to detect significant numbers of cells expressing the T-cell lineage markers CD4 and CD8.
Western blot analysis of bone marrow cells of wild-type and Shb knockout leukemic mice was performed in order to investigate the effects of Shb deletion on the activation levels of signaling intermediates that have been established as Shb-regulated targets. 5 Activation of FAK and AKT was unaffected by the absence of Shb, whereas the phosphorylation of activating sites of ERK was significantly elevated in the absence of Shb (Figure 4(a)). Bone marrow cells were c-Kit+ enriched to study the expression levels of certain cytokines. The transcription of Csf3, Csf2, Il-3, and Il-6 were similar in wild-type and knockout bone marrows, whereas Il-7 and Cxcl-12 showed significantly lower expression levels in the absence of Shb (Figure 4(b)). Therefore, the elevated number of WBCs in wild-type transplanted mice might be related to the expression levels of these cytokines.

Signal transduction and cytokine expression in p190 BCR-ABL leukemia due to the absence of Shb. (a) FAK, ERK, and AKT activation. Their phosphorylation levels are shown in the total bone marrow from leukemic mice receiving wild-type and Shb knockout cells expressing p190 BCR-ABL, by western blot analysis of the same wild-type and knockout tumor samples. Signal strength was evaluated by densitometric analysis and related to total ERK, total FAK, and total AKT as indicated. Values are presented as arbitrary units ± SEM, based on 10 mice from each genotype in three independent experiments. (b) Expression levels of cytokines in c-Kit+ bone marrow cells in p190 BCR-ABL-transplanted mice by semi-quantitative real-time RT-PCR. The CT values were normalized by subtracting the corresponding β-actin values. Shb knockout cells were related to equivalent wild-type values. The expression differences were then determined as 2–ΔKOCt–WTCt ± SEM to identify fold change in expression level. Values are based on c-Kit+ cells from three mice of each genotype in one experiment. * indicates p < 0.05 as confirmed by Student’s t test.
Lymph node engagement has been demonstrated in p190 BCR-ABL leukemia. More leukemic mice receiving transduced wild-type bone marrow displayed a detectable limp in their movement (8 of 13) than Shb-null recipients (3 of 13; p < 0.05), probably due to enlarged lymph nodes. Immunostaining of enlarged lymph nodes from wild-type mice revealed the abundant presence of cells expressing B220 that stained with variable intensity, showing invasion of B lymphocytes to the lymph nodes (Supplementary Material Figure 1(c)). These data further support the notion that overt B-cell leukemia with lymph node infiltration prevails in the wild-type scenario, whereas it appears to be a less prominent feature in the Shb knockout situation.
Shb deficiency increases thymus size in KrasG12D-induced T-cell leukemia/thymic lymphoma
The Kras2LSL/Mx1-Cre (KM) mouse model has been used for triggering the expression of an oncogenic KrasG12D in the bone marrow allowing the mice to develop T-cell leukemia and/or T-cell thymic lymphoma.31,32 A combination of this oncogene with Shb deletion was used in order to investigate the effects of Shb deficiency on Kras-induced T-cell leukemia.
The survival period after transplantation of wild-type or Shb knockout KrasG12D bone marrow cells to wild-type recipients failed to show any significant difference by log-rank test (p = 0.10; Figure 5(a)). The size of the liver and spleen as well as weight gain remained unchanged between the genotypes, whereas the thymus size was significantly larger in Shb-null transplanted mice indicating a more severe degree of thymic lymphoma (Figure 5(b)). In mice carrying the Kras oncogene on a wild-type background, 5 of 13 mice had an enlarged thymus and elevated WBC (>15 million WBCs/mL). The corresponding numbers in the knockout situation were 9 and 7, respectively, of 12 mice. The numbers of bone marrow cells, WBCs, RBCs and platelets were not significantly different between the genotypes (Figure 5(c)–(f)). Shb knockout WBC counts, however, showed an extreme degree of variation between the mice with values ranging from 0 and 243 million WBC/mL blood. The presence of B-lymphocytes, myeloid cells, T-lymphocytes, and lineage markers in peripheral blood (Figure 5(g)) and bone marrow cells (Fig. 5(h)) as determined by flow cytometry was found to be the same in wild-type and Shb knockout mice.

Absence of Shb- and Kras-induced T-cell leukemia/thymic lymphoma. (a) Kaplan–Meier survival analysis of Kras-induced T-cell leukemia analyzed by log-rank test of mice transplanted with wild-type and Shb-null KrasG12D bone marrow cells. By Student’s t test, due to normal distribution of mean survival time in each experiment, shorter latency can be noted in Shb-deficient cohort (p < 0.05). (b) The liver, spleen, and thymus weights as well as the amount of gained weight when moribund. (c) Number of bone marrow cells of the iliac, tibia, and femur bones of recipient mice at the time of death. (d)–(f) Analysis of blood profile, indicating the numbers of WBCs, RBCs, and platelets. FACS analysis of lineage-defining marker-expressing cells including B-cell, myeloid, and T-cell as well as lineage+ cells in (g) peripheral blood and (h) bone marrow of mice receiving wild-type and Shb knockout KrasG12D-expressing bone marrow cells. Results are presented as mean values ± SEM from 13 or 11 mice of each genotype in four independent experiments. * denotes p < 0.05 as determined by Student’s t test.
Bone marrow activation of FAK, ERK, and AKT was unaffected by the absence of Shb (Supplementary Material Figure 3). In addition, there was no effect on the bone marrow cytokine expression (Supplementary Material Figure 4).
The increased severity of thymic lymphoma as a consequence of Shb deficiency prompted us to investigate T-cell development in the thymus. It is known that T-lymphocyte progenitors are raised in the bone marrow and migrate to the thymus where they initially do not express TCR, CD4, and CD8 (double negative (DN)). As these cells migrate through the thymus, they will mature and eventually express TCR, CD4, and CD8 (double positive (DP)). Further differentiation leads to single positive (SP) CD4 and CD8 cells, of which the former become T-helper cells and the latter cytotoxic T cells. 33 Staining the thymus for these T-cell markers showed a higher content of CD4/CD8 (DP) cells in Shb KO KrasG12D recipients, while CD8 (SP) were simultaneously decreased (Figure 6). No differences in the DN1-4 populations were noted (Supplementary Material Figure 5). Consequently, the primary site of SHB’s action in this experimental model appears to be related to later stages of T-cell development in the thymus causing increased severity of thymic lymphomas.

T-cell development in the thymus in the absence of Shb in Kras-induced T-cell leukemia/thymic lymphoma. FACS analysis of T-cell markers (CD4/CD8) within thymocytes of T-cell leukemic mice. The results are presented as mean values ± SEM from six mice of each genotype in three independent experiments. * represents p < 0.05 as determined by Student’s t test.
Discussion
The study demonstrates altered disease characteristics in experimental models of CNL, B-cell leukemia, and T-cell leukemia/thymic lymphoma as a consequence of Shb deficiency. The effects were pleiotropic and specific for each disease model. The most plausible explanation is that the differences in SHB signaling modulate the phenotype of the tumor cell and that this may have consequences for the progression of disease.
In this study, the effects of Shb deficiency in CSF3RT618I-induced leukemia were investigated. As a result, shorter latency and an elevated level of WBCs in peripheral blood in Shb-null recipients suggested a more aggressive disease in this model. Moreover, FACS analysis of peripheral blood displayed the presence of more mature myeloid cells as well as total lineage+ cells in Shb knockout recipients, suggesting increased egress of relatively mature myeloid cells from the bone marrow into blood. Since the CSF3RT618I mutation operates in a ligand-independent manner, there is no need for the granulocyte colony stimulating factor (G-CSF) ligand to activate the receptor. It has been shown previously that the absence of Shb in p210 BCR-ABL-induced myeloid leukemia results in elevated FAK activity. 9 This effect was paralleled by neutrophilia and increased expression levels of IL-6 and G-CSF. 5 In the current setting, the absence of Shb alters the signaling characteristics that in cooperation with the T618I membrane proximal mutation cause a decrease in the expression of IL-6 and G-CSF compared to wild-type T618I-expressing bone marrows. Despite a reduction in cytokine production, the changed signaling signature due to Shb deficiency including the activation of FAK and ERK will result in the accumulation of relatively mature leukocytes in peripheral blood. This explains why a more aggressive disease can be observed in the absence of Shb.
Shb deficiency displayed important differences in the p190 BCR-ABL B-cell ALL model compared with p210 BCR-ABL myeloid leukemia. One divergence was that currently no further FAK activation could be detected, which in the p210 case was considered to be of prime significance for the phenotype. However, increased activation of ERK was observed. It has previously been demonstrated that BCR-ABL activates the Ras/Raf/MEK/ERK, JAK/STAT, and phosphatidylinositide 3-kinase/AKT signal transduction pathways, resulting in enhanced proliferation. 29 In addition, the expression of the cytokines IL-7 and chemokine C-X-C motif ligand 12 (CXCL-12) was reduced. IL-7 is an essential factor for growth, differentiation, and survival of B-cell precursors. 34 Development of B cells, starting from HSCs to immature B cells, occurs in the bone marrow and the last step of maturation, subsequent to immature B cells, happens in peripheral blood. 35 By a reduction of bone marrow IL-7 expression, a reduction in leukemic cell proliferation is anticipated. CXCL-12 is important for HSC and bone marrow progenitor cell retention in the bone marrow36,37 and consequently increased egress of such cells is expected. A reduction of these cytokines may in concert explain the observed WBC pattern; lower WBC counts due to less IL-7 stimulated the proliferation and more immunophenotypically mature WBC due to increased egress from the bone marrow compartment. It still remains a conundrum why latency is unchanged despite fewer and more mature WBCs at the time of death but presumably this reflects altered blood cell characteristics that have deleterious effects on survival. There is also a higher percentage of mature neutrophils present in blood in the absence of Shb, as previously shown in the p210 BCR-ABL leukemia model. Apparently, this is a common feature of Shb deficiency in both models of BCR-ABL-dependent leukemia, further linking SHB function to the BCR-ABL oncogene in CNL. Enhanced ERK signaling has been shown to be essential for CSF3R-induced CNL, 38 and since increased ERK activity was observed in these experimental models, due to the absence of Shb, this is likely the common feature explaining neutrophilia. We have demonstrated that SHB and PAX5 are commonly co-expressed in patients with AML. 6 These two genes were instrumental in defining a subgroup of AML with immune cell characteristics, 6 in agreement with the fact that Pax5 is important to reach a B-cell committed status by adjusting lineage-specific genes. 39 SHB may thus operate at an interface between different forms of leukemia, shifting the phenotypic characteristics of multi-potent leukemic cells.
It is known that the expression of oncogenic Kras alone is not sufficient to initiate leukemogenesis and that other genetic changes are required as well. It seems that there is a first genetic hit, which is oncogenic Kras, that occurs in the bone marrow on HSCs and a second genetic hit that occurs on T-cell progenitors, which can be detected in the thymus. 40 We find no evidence for alterations in the bone marrow as a consequence of Shb deficiency but the data rather suggest effects on late T-cell development in the thymus. Consequently, Shb deficiency resulted in a more aggressive form of Kras-induced T-cell thymic lymphoma with an increase of the thymus size. In order to become functional T cells, lymphoid progenitors undergo developmental steps via important signaling pathways, including TCR signaling.41,42 Some key elements in this signaling pathway are the adaptors lymphocyte cytosolic protein 2 (SLP76)43,44 and linker for activation of T cells (LAT).45,46 SHB has been shown to associate with TCR, LAT, and SLP76 and consequently plays a critical role in TCR signal transduction,1,4 that is, the absence of Shb results in an elevated proliferative response to TCR stimulation. 8 Shb-null thymocytes displayed an increased level of CD8/CD4 DP cells, indicating effects on later stages of T-cell development of relevance for the expansion of the thymus size. This is in line with the previously described participation of SHB in TCR signaling and T-cell maturation/proliferation.1,8,10
Conclusion
Disparate effects on Shb deficiency presently are described related to multiple sites of action in three models of leukemia. Leukemia is a multi-facetted disease and different forms of this disease exhibit shared interfaces/genetic aberrations. The data suggest that SHB operates at such interfaces shifting phenotypic responses, thus explaining the pleiotropic nature of the responses to Shb deficiency. The data help explain the characteristics of human AML with high SHB mRNA, which was associated with immune cell characteristics and unfavorable prognosis.
Supplemental Material
Legends_to_supplemental_figures_TUB – Supplemental material for Disparate effects of Shb gene deficiency on disease characteristics in murine models of myeloid, B-cell, and T-cell leukemia
Supplemental material, Legends_to_supplemental_figures_TUB for Disparate effects of Shb gene deficiency on disease characteristics in murine models of myeloid, B-cell, and T-cell leukemia by Maria Jamalpour, Xiujuan Li, Karin Gustafsson, Jeffrey W Tyner and Michael Welsh in Tumor Biology
Footnotes
Acknowledgements
We are grateful to Dr George Daley for suggestions, to Dr Martin Bergö for suggestions and providing the mutant Kras mice, and to Anna Bereza-Jarocinska for help with qPCR and western blotting. Maria Jamalpour, Xiujuan Li, Karin Gustafsson, and Michael Welsh performed the experiments. Maria Jamalpour, Karin Gustafsson, Jeffrey W Tyner, and Michael Welsh planned the experiments. All authors interpreted the results and Maria Jamalpour and Michael Welsh wrote the paper. All authors have made comments to the text.
Supplementary materials
The supplementary materials supporting the conclusions of this article are included within the article and its online additional files.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Swedish Cancer Foundation, The Swedish Research Council, EXODIAB, and the Family Ernfors Fund. J.W.T. was supported by grants from the V Foundation for Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and the NCI (5R00CA151457-04 and 1R01CA183947-01).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.