
Abstract
Background:
Acute myeloid leukemia (AML) is an extremely heterogeneous malignant disorder; AML has been reported as one of the main causes of death in children. The objective of this work was to classify the most deleterious mutation in CCAAT/enhancer-binding protein-alpha (CEBPA) and to predict their influence on the functional, structural, and expression levels by various Bioinformatics analysis tools.
Methods:
The single nucleotide polymorphisms (SNPs) were claimed from the National Center for Biotechnology Information (NCBI) database and then submitted into various functional analysis tools, which were done to predict the influence of each SNP, followed by structural analysis of modeled protein followed by predicting the mutation effect on energy stability; the most damaging mutations were chosen for additional investigation by Mutation3D, Project hope, ConSurf, BioEdit, and UCSF Chimera tools.
Results:
A total of 5 mutations out of 248 were likely to be responsible for the structural and functional variations in CEBPA protein, whereas in the 3′-untranslated region (3′-UTR) the result showed that among 350 SNPs in the 3′-UTR of CEBPA gene, about 11 SNPs were predicted. Among these 11 SNPs, 65 alleles disrupted a conserved miRNA site and 22 derived alleles created a new site of miRNA.
Conclusions:
In this study, the impact of functional mutations in the CEBPA gene was investigated through different bioinformatics analysis techniques, which determined that R339W, R288P, N292S, N292T, and D63N are pathogenic mutations that have a possible functional and structural influence, therefore, could be used as genetic biomarkers and may assist in genetic studies with a special consideration of the large heterogeneity of AML.
Introduction
Acute myeloid leukemia (AML) is an extremely heterogeneous malignant disorder; in recent years, AML developed so rapidly by affecting children and adults, and hence it has been reported as one of the main causes of death in children.1-5 It is the most common acute leukemia in adults,6-8 with a frequency of more than 20 000 cases per year in the United States alone. 6 It is characterized by genetic alterations in hematopoietic ancestor cells that change usual mechanisms of self-replicating.2,9 AML is commonly triggered by mutation in CCAAT/enhancer-binding protein-alpha (CEBPA) gene.10-14 The CEBPA is a transcription element that affects immune cell density and diversity.15,16 Most patients with AML who have CEBPA alterations simultaneously transport double mutations,17-19 nevertheless, different mutations have been reported20-24; some studies have been reported which claimed some related factors besides CEBPA mutation, such as smoking, alcohol, and exposure to solvents and agrochemicals may cause AML,25-27 but no evidence of publication bias. Other genes have been reported which cause AML such as fms-related tyrosine kinase 3 (FLT3-ITD) and nucleophosmin 1 (NMP1), which assisted to improve person diagnosis; furthermore, these mutant molecules characterize as a potential target for molecular therapies.6,28-31 Sometimes, patients with chronic lymphocytic leukemia can develop AML, 32 whereas in rare cases patients with AML can develop esophageal cancer. 33
Stem cell transplantation treatment is related to the result of treatment for patients with cytogenetically usual AML. 34 Nevertheless, the advantage of the transplant is exclusive to subcategory of patients with CEBPA mutations alone31,34; in spite of this hopeful recent evolution, the outcomes of patients with AML remain insufficient, with more than 50% of the patients eventually dying from this devastating disease. The purpose of this study is to classify functional mutations located in the coding region of CEBPA gene using in silico analysis.
Disease-causing single nucleotide polymorphisms (SNPs) are frequently found to arise at evolutionarily conserved regions; these have a key role at structural and functional levels of the protein. The capability to calculate whether a particular SNP is deleterious or not is very important for the prognosis of disorder.35-45 The practice of translational bioinformatics has solid influence on the identification of candidate SNPs and can contribute in pharmacogenomics by identifying high-risk SNP mutation contributing to drug response as well as developing novel therapeutic elements for this deadly disease.46-54 This is the first silico analysis in coding and non-coding regions of CEBPA gene that prioritized SNPs to be used as diagnostic markers with a special consideration of the large heterogeneity of AML among different populations.
Materials and Methods
Data mining
The polymorphic data of CEBPA gene were claimed from National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/), and the reference sequence of human protein was collected from UniProt 55 (https://www.uniprot.org/).
Functional analysis
Sorting Intolerant From Tolerant
It is the first in silico functional analysis that calculates whether an amino acid alteration affects protein function or not. Sorting Intolerant From Tolerant (SIFT) scores < 0.05 are expected to be damaging altered amino acid, otherwise it is considered to be tolerant. 56 It is available at https://sift.bii.a-star.edu.sg/.
PolyPhen-2
It is a trained machine learning to predict whether an amino acid replacement affects protein function and structure or not, by calculating position-specific independent count (PSIC) for each SNP at a time. There are 2 outputs whether probably damaging (values are more frequently 1) and possibly damaging or benign (values range from 0 to 0.95). 57 It is available at http://genetics.bwh.harvard.edu/pph2/.
PROVEAN
It is an online in silico functional analysis tool that calculates whether an amino acid replacement has an influence on the organic function of a protein stranded on the alignment-based score. If the PROVEAN score ⩽ –2.5, the protein variant is expected to have a “deleterious” effect, whereas if the PROVEAN score is >–2.5, the variant is expected to have a “neutral” effect. 58 It is available at http://provean.jcvi.org/index.php.
SNAP2
It is a trained functional analysis tool that differentiates between effect and neutral SNPs by taking various features into validation. SNAP2 got an accuracy of 83%, which has 2 expectations: effect (positive score) or neutral (negative score). It is considered an important and substantial enhancement over other methods. It is available at https://rostlab.org/services/snap2web/.
SNPs&GO
It is a trained machine learning based on the technique to precisely calculate the deleterious associated alterations from protein sequence. SNPs&GO collects in a unique framework information derived from protein sequence, evolutionary information, and function as coded in the Gene Ontology terms and underperforms other available predictive methods (PhD-SNP and PANTHER). 59 It is available at http://snps.biofold.org/snps-and-go/snps-and-go.html.
PMut
It is a web-based tool for the explanation of SNP alternates on proteins, which allows the rapid and precise calculation (80%) of the compulsive features of each SNP grounded on the practice of neural networks. 60 It is accessible at http://mmb.irbbarcelona.org/PMut.
Stability analysis
I-Mutant 3.0
I-Mutant is a support vector machine (SVM)-based tool. I-Mutant predicts whether the protein mutation stabilizes or destabilizes the protein structure by calculating free energy change by coupling predictions with the energy-based FOLD-X tool. 61 It is available at http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi.
MUpro
It is a structural analysis online tool for the calculation of protein stability variations in arbitrary SNPs. The value of the energy change is expected, and assurance mark between −1 and 1 for evaluating the assurance of the expectation is calculated. A score of <0 means the mutant decreases the protein stability; conversely, a score of >0 means the mutant increases the protein stability. 62 It is available at http://mupro.proteomics.ics.uci.edu/.
3-Dimensional clustering analysis
Mutation3D
It is a functional calculation and visualization online tool for investigating the 3-dimensional (3D) plan of amino acid alterations in protein models and structures. 63 It is available at http://mutation3d.org.
Biophysical validation
Project HOPE
It is a web server to search protein 3D structures by bringing together structural information from several sources such as UniProt database. The main aims for the submissions in Project HOPE are to analyze and confirm the results that we obtained earlier. It is available at http://www.cmbi.ru.nl/hope.
Conservational analysis
BioEdit
It is a software package proposed to stream a distinct program that can run nearly any sequence operation as well as a few basic alignment investigations. It is available for download at http://www.mbio.ncsu.edu/bioedit/bioedit.html.
ConSurf server
It is a web server that offers evolutionary conservation summaries for proteins of known structure in the protein data bank. ConSurf spots the parallel amino acid sequences and runs multialignment methods. The conserved amino acid across species flags its position using specific algorithm. 64 It is available at http://consurf.tau.ac.il/.
3D structural analysis
RaptorX
The 3D structure of human CEBPA protein is not available in the Protein Data Bank. Hence, RaptorX was used to make a 3D structural model for wild-type CEBPA. RaptorX is a web server predicting structure property of a protein sequence without using any templates. 65 It is available at http://raptorx.uchicago.edu/.
UCSF Chimera
It is a visualization analysis program of 3D structure prototype, docking analysis, and so many related analyses. A predicted model was created by RaptorX to visualize and compare the amino acid alterations using UCSF Chimera. 66 UCSF Chimera 1.8 is free for download at http://www.cgl.ucsf.edu/chimera/.
GeneMANIA
It is a method to know protein function prediction integrating multiple genomics and proteomics data sources to make inferences about the function of unknown proteins. 67 It is available at http://www.genemania.org/.
Variant Effect Predictor
The Ensembl Variant Effect Predictor (VEP) software provides tools and methods for a systematic approach to annotate and aid prioritization of variants in both large-scale sequencing projects and smaller analysis studies. 68 It is available at http://www.ensembl.org/vep.
PolymiRTS server
It is a server for investigating functional SNPs in 3′-untranslated region (3′-UTR) of CEBPA gene that may change miRNA binding on target sites, resulting in different functional consequences. 69 It is available at http://compbio.uthsc.edu/miRSNP/.
Results
The total number of SNPs in different regions of CEBPA gene was retrieved from NCBI. The distribution of non-synonymous single nucleotide polymorphisms (nsSNPs) in coding and non-coding regions of CEBPA gene contained 248 nsSNPs, with 350 SNPs in the 3′-UTR and 11 in the 5′-untranslated region (5′-UTR; Figure 1).

The distribution of SNPs in coding and in non-coding regions of CEBPA gene. SNPs indicates single nucleotide polymorphisms.
A total of 248 missense mutations were retrieved from the database of single nucleotide polymorphism (dbSNP)/NCBI database, and these SNPs were submitted into different functional analysis tools such as SIFT, polymorphism phenotyping v2 (PolyPhen-2), PROVEAN, and SNAP2, respectively. Sorting Intolerant From Tolerant server predicted 28 deleterious SNPs, PolyPhen-2 predicted 85 damaging SNPs (29 were possibly damaging and 56 were probably damaging to protein), PROVEAN represented 34 deleterious SNPs, whereas in SNAP2 we filtered the triple-positive deleterious SNPs from the previous 3 analysis tools, out of 53 SNPs there were 19 predicted deleterious SNPs by SNAP2. Table 1 represents the Quad-positive of deleterious SNPs after filtrations, the number decreased rapidly to 19 SNPs, after submitting them into SNPs&GO and PhD-SNP, PMut and PANTHER, respectively, to run more investigation on these SNPs and their effect on the functional level. The triple positive in the 3 tools was 5 disease-associated SNPs (Table 2). Finally, we submitted them to I-mutant 3.0 and MUpro, respectively, to investigate their effect on structural level. The 2 online tools revealed that all 5 mutations predicted a dramatic decrease in the protein stability, except for 2 SNPs (N292T and D63N) that were predicted by I-Mutant to increase the stability of the protein (Table 3).
Affect or damaging nsSNPs associated predicted by various software.
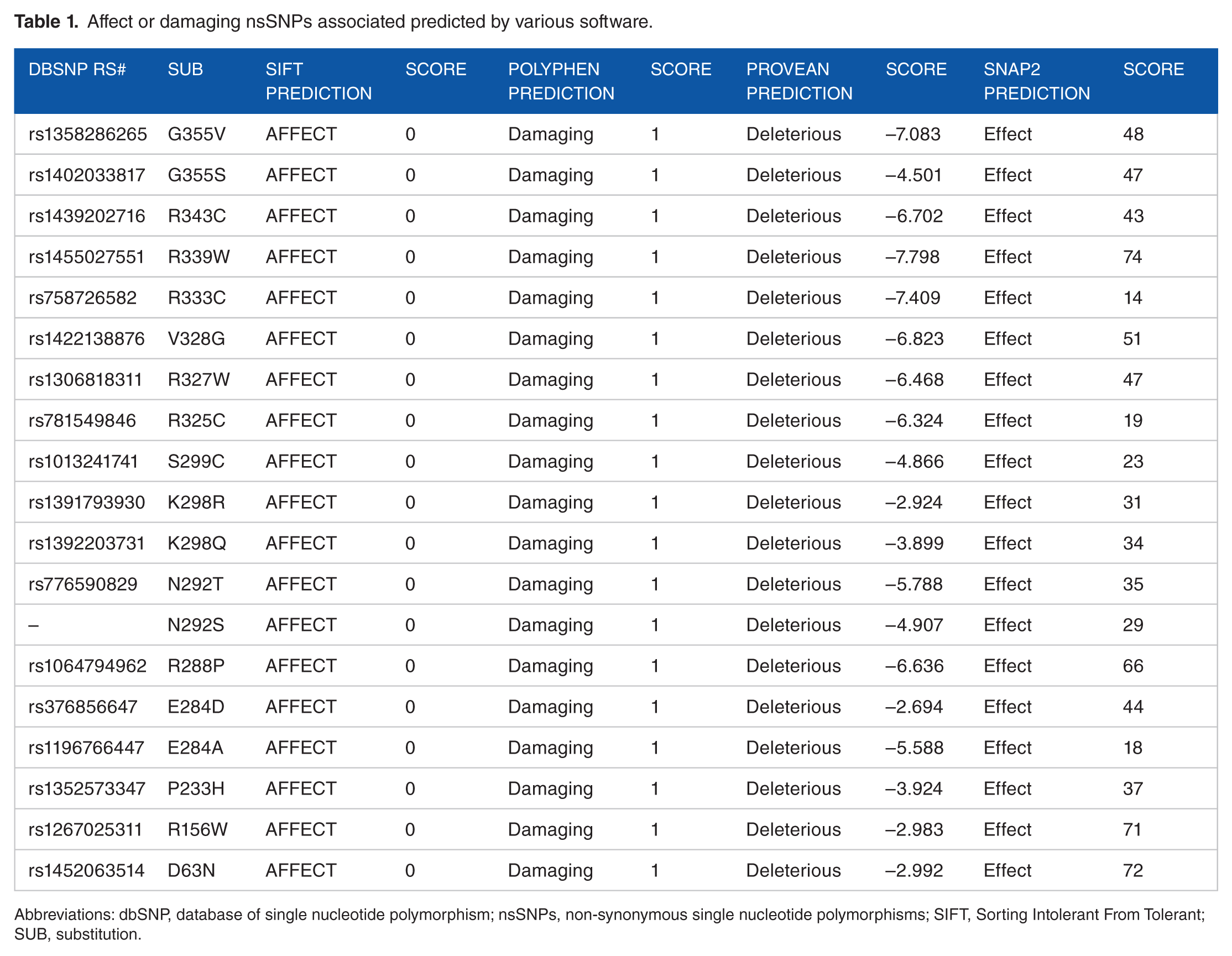
Abbreviations: dbSNP, database of single nucleotide polymorphism; nsSNPs, non-synonymous single nucleotide polymorphisms; SIFT, Sorting Intolerant From Tolerant; SUB, substitution.
Pathogenic nsSNPs associated variations predicted by various software.

Abbreviations: nsSNPs, non-synonymous single nucleotide polymorphisms; RI, reliability index; SUB, substitution.
Structural investigation calculated using I-Mutant 3.0 and MUpro.

Abbreviations: dbSNP, database of single nucleotide polymorphism; RI, reliability index; SUB, substitution.
DDG value: free energy changes value.
Single nucleotide polymorphisms in 3′-UTR of CEBPA gene were submitted as batch to PolymiRTS server. The result shows that among 350 SNPs in the 3′-UTR of CEBPA gene, about 11 SNPs were predicted, namely, rs116528776, rs113670631, rs34017519, rs146104564, rs187516157, rs2376497, rs192371350, rs1049969, rs41367646, rs184965384, and rs187751931; among these 11 SNPs, 65 alleles disrupted a conserved miRNA site and 22 derived alleles created a new site of miRNA (Table 6).
Discussion
In vitro mutagenesis, functional and characterization studies, is an unwieldy task regarding workload, time, and fees. For these reasons, bioinformatics analysis is an appropriate, rapid, low-cost, and dependable approach to enhance our understanding of how mutations could disturb the protein structure and function.53,54 Disease-causing SNPs are commonly found to arise at evolutionarily conserved regions. Those have a key role at structural and functional levels of the protein36,38; therefore, our focus was dedicated to the coding region, which unmasked 5 mutations in CEBPA gene using different sequence and structure-based algorithms (Figure 2). The SNPs that have been found in this study could be used in prognostics of disease, because identification of CEBPA status in AML has a major clinical importance, allowing relapse risk to be stratified properly for post-remission treatment.70,71

Descriptive workflow of softwares used in SNP analysis. SNP indicates single nucleotide polymorphism.
All these SNPs (D63N, R288P, N292T, N292S, and R339W) were retrieved from the dbSNPs/NCBI database as untested and all were found to be pathogenic mutations.
At the functional level analysis, our results showed that all these nsSNP substitutions (D63N, R288P, N292T, N292S, and R339W) were classified as highly pathogenic mutations (Table 1). The analysis of different SNPs on the protein structure can disturb interactions with other molecules, MUpro results showed a decrease in stability for all these SNPs (D63N, R288P, N292T, N292S, and R339W), whereas I-Mutant results showed a decrease in stability for these SNPs (R288P, N292S, and R339W), thus suggesting that these mutations could directly or indirectly destabilize the amino acid interactions triggering functional deviations of protein to some point.
CEBPA offers information for building a protein termed CCAAT enhancer-binding protein alpha. It’s a transcription factor (TF) and its performance is a malignant suppressor, which means it is complicated in cellular mechanisms and could help to prevent the cells from developing and dividing too swiftly or in an uncontrolled mode and that is the principle of cancer.24,72 We also achieved analysis by Mutation3D server, all SNPs in red (R288P, N292S, and N292T) are clustered mutation, significantly, such mutation clusters are commonly associated with human cancers, 73 whereas SNPs in blue (R339W) and gray (D63N) are covered and uncovered mutations, respectively (Figure 3).

Structural simulations for mutant residues in CEBPA protein, demonstrated by Mutation3D.
Project HOPE server was used to submit the most damaging SNPs (R288P): interestingly, proline interrupts an α-helix when not positioned at 1 of the first 3 positions of that helix. If this happened, a major impact on the protein structure could occur (Figure 4). In this study, we also observed that only 1 SNP (D63N), the residue predicted to be mutated, is evolutionarily conserved across species, and this may increase the possibility of altered transcriptional and cell cycle regulation (Figure 5).

The mutant residue located in an α-helix.

Alignments of 8 amino acid sequences of CEBPA representing that the residues predicted to be mutated are evolutionarily conserved across species. Sequences alignment was done by BioEdit (v7.2.5).
The 3D protein structure analysis enables mapping of amino acid substitutions and, therefore, RaptorX was used to make a 3D structure model for CEBPA protein (Figure 6) to support and match the results acquired from different computational tools, UCSF Chimera serves this purpose (Figures 7 to 11), show the differences between native and mutant amino acids, in the green and red boxes the schematic structures of the native amino acids (in the left side), and the mutant ones (in the right side). The backbone, which is the same for each amino acid, is colored red and the side chain, unique for each amino acid, is colored black, the 3D wide-type residues colored green and mutant ones colored red, whereas the protein is colored cyan.

The 3D structure of the CEBPA protein model was generated by using RaptorX; it could not generate the 3D structure of all amino acid positions; therefore, the model was done from positions 52 to 358, due to the lack of information. 3D indicates 3-dimensional.

D63N: aspartate (green box) changes to asparagine (red box) at position 63.
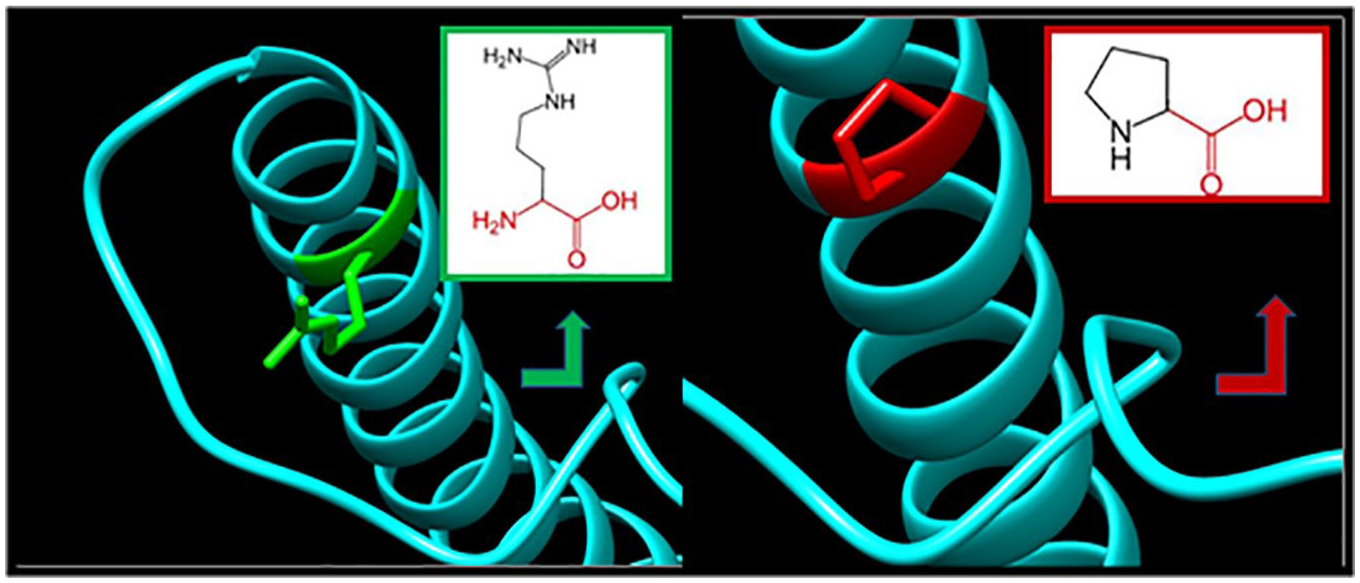
R288P: the amino acid arginine changes to proline at position 288; illustration was done by UCSF Chimera (v 1.8.) and project HOPE.

N292S: the amino acid asparagine changes to serine at position 292; illustration was done by UCSF Chimera (v 1.8.) and project HOPE.

N292T: the amino acid asparagine changes to threonine at position 292; illustration was done by UCSF Chimera (v 1.8.) and project HOPE.

R339W: the amino acid arginine changes to tryptophan at position 339; illustration was done by UCSF Chimera (v 1.8.) and project HOPE.
In Figure 7, D63N shows the native amino acid (aspartic acid) and the mutant one (asparagine) at position 63; the mutated residue is located on the surface of a domain with unknown function; the residue was not found to be in contact with other domains of which the function is known within the used structure; however, contact with other molecules or domains is still possible and might be affected by this mutation.
In Figure 8, R288P shows close-up angle of the native amino acid (arginine) and the mutant one (proline) at position 288; the mutated residue is located in a domain that is important for the activity of the protein and in contact with residues in another domain, and it is possible that this interaction is important for the correct function of the protein. The mutation can affect this interaction and as such affect protein function; the mutation introduces an amino acid with different properties, which can disturb this domain and abolish its function, the charge of the wild-type residue is lost by this mutation, which can cause loss of interactions with other molecules; the mutant residue is smaller than the wild-type residue; and this will cause a possible loss of external interactions.
In Figure 9, N292S shows the schematic structures of the original amino acid (asparagine) and the mutant one (serine) at position 292; each amino acid has its own specific size, charge, and hydrophobicity value. The original wild-type residue and newly introduced mutant residue often differ in these properties; the mutant residue is more hydrophobic than the wild-type residue; the mutant residue is smaller than the wild-type residue; and this will cause a possible loss of external interactions.
In Figure 10, N292T shows close-up angle of the native amino acid (asparagine) and the mutant one (threonine) at position 292; the mutated residue is located in a domain that is important for the activity of the protein and in contact with residues in another domain. It is possible that this interaction is important for the correct function of the protein. The mutation can affect this interaction and as such affect protein function; the mutant residue is smaller than the wild-type residue; and this will cause a possible loss of external interactions.
In Figure 11, R339W shows the schematic structures of the original amino acid (arginine) and the mutant one (tryptophan) at position 339; the residue is located on the surface of the protein; mutation of this residue can disturb interactions with other molecules or other parts of the protein; and the charge of the wild-type residue (positive) is lost by this mutation. This can cause loss of interactions with other molecules. The mutant residue is more hydrophobic than the wild-type residue, which can disturb this domain and abolish its function.
We also used ConSurf web server; the nsSNPs that are shown by black boxes located in highly conserved regions and predicted to cause structural and functional impacts on CEBPA protein (Figure 12).

The conserved amino acids across species in CEBPA protein were determined using ConSurf. e: exposed residues according to the neural-network algorithm are indicated in orange letters. b: residues predicted to be buried are demonstrated via green letters. f: predicted functional residues (highly conserved and exposed) are indicated with red letters. s: predicted structural residues (highly conserved and buried) are demonstrated in blue letters. I: insufficient data—the calculation for this site performed in less than 10% of the sequences is demonstrated in yellow letters.
GeneMANIA revealed strong functional associations that CEBPA gene had observed with transforming growth factor beta (TGFB1) and tumor necrosis factor (TNF) genes (Figure 13). Besides, weak interactions with less confidence have been observed for prolactin regulatory element binding (PREB) and early B cell factor 1 (EBF1) genes. The genes co-expressed with, sharing similar protein domain, or contributed to achieve similar function are shown in Tables 4 and 5.

Interaction between CEBPA and its related genes.
The CEBPA gene functions and its appearance in network and genome.
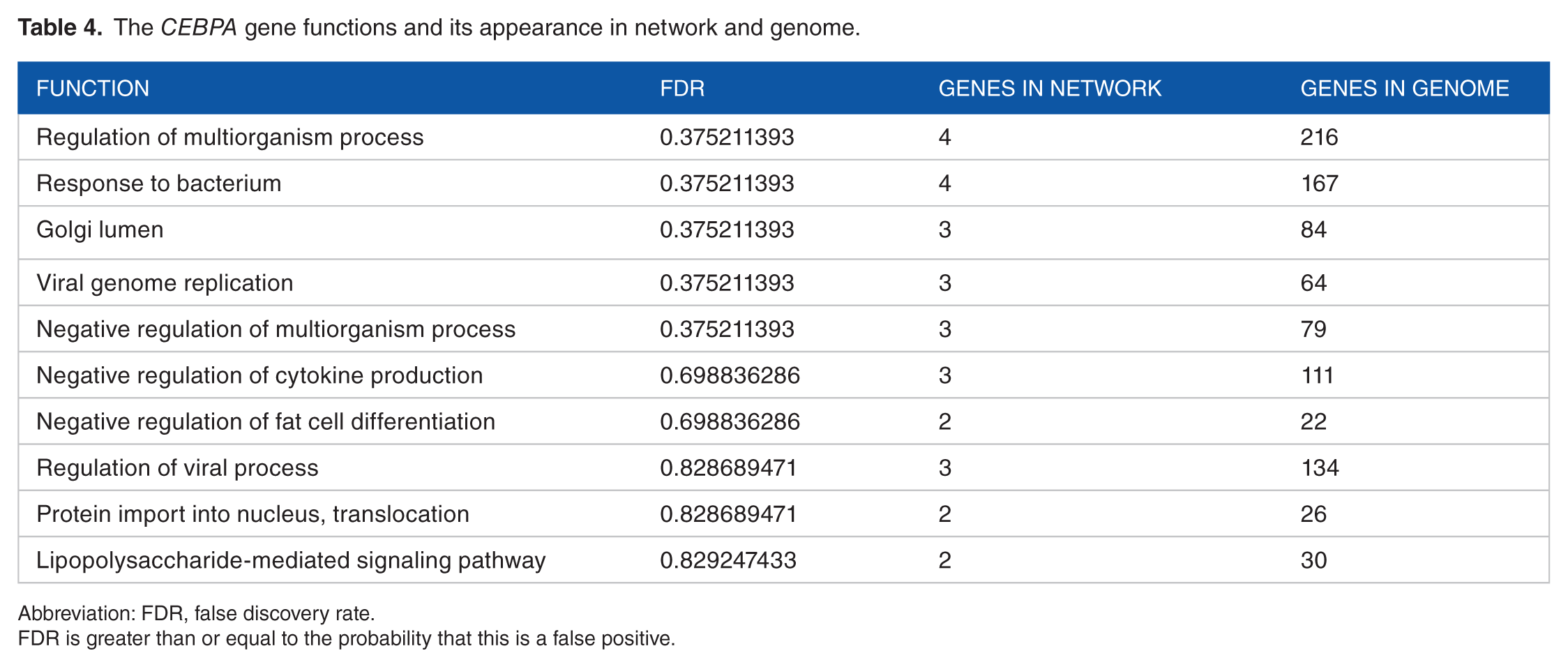
Abbreviation: FDR, false discovery rate.
FDR is greater than or equal to the probability that this is a false positive.
The gene co-expression, shared domain, and interaction with CEBPA gene network.

The VEP annotates variants using a wide range of reference data, including transcripts, regulatory regions, and frequencies from previously observed variants, citations, clinical significance information, and predictions of biophysical consequences of variants, and that is what makes VEP to give accurate results; as far as we know, the only limitation is that VEP annotates each input variant independently, without considering the potential compound effects of combining alternate alleles across multiple variant loci, 68 and this is the reason why we could not predict the consequences of N292S mutation, whereas the predicted variant consequences are shown in Table 6. VEP reported regulatory consequences for many variants, including 5 variants within a coding region, 6 variants within a non-coding region, 10 variants within upstream gene, 6 variants within downstream gene, 4 variants within non-coding transcript exon, and 1 variant within transcription factor binding site (TFBS); in conclusion, mutations within a coding region affect the protein function, whereas regulatory variants within non-coding genomic regions can greatly affect disease and could be involved in the specific recruitment or sequestration of spliceosome factors and RNA-binding proteins (RBPs)74,75; the SNPs in the upstream, downstream, 5′-, and 3′-UTRs might affect transcription or translation process 76 ; whereas alteration at TFBS has many consequences, such as variants within a TFBS differentially influence its TF-binding affinity; another consequence that could affect TFBS is that multiple variants in the promoter regions can “transform” an existing binding site of a particular TF into a site for another TF, even from a different TF family. 77 Figure 14 illustrates the summary pie charts and statistics.
Shows variant consequences, transcripts, and regulatory features by VEP tool.

Abbreviations: TF, transcription factor; VEP, Variant Effect Predictor.

Summary pie charts and statistics.
Single nucleotide polymorphisms in 3′-UTR of CEBPA gene were submitted as batch to PolymiRTS server. The result showed that 11 SNPs may affect microRNA binding sites. As an example, rs2376497 SNP containing (D) allele had 8 microRNA sites (miRSite) as target binding site that can disrupt a conserved miRNA and (C) alleles had 5 miRSites that disrupt a conserved miRSite. Table 7 demonstrates the SNPs predicted by PolymiRTS to induce disruption or formation of miRSite.
SNPs and INDELs in miRNA target sites in CEBPA gene.

Abbreviations: miRSite, microRNA site; SNPs, single nucleotide polymorphisms.
D: the derived allele disrupts a conserved miRNA site (ancestral allele with support ⩾ 2). C: the derived allele creates a new miRNA site.
The limitations of this study are that it focuses on coding and 3′-UTRs using different numbers of tools of silico analysis; yet there are number of genes responsible for AML although AML is frequently triggered by mutation in CEBPA gene10-13; in general, it is likely to achieve that computational approach remains as an accurate way to make a rapid analysis regarding the expected effect of mutations; nevertheless, the more factors that are taken into account, the more accurate the prediction will be. To take the best advantage of bioinformatics analysis, different computational tools could be used, trying to cover the major aspects influencing protein structure and function, Mutation Taster, 78 SNPdryad, 79 and ACES (a machine learning toolbox for clustering analysis and visualization). 80 The 5′-UTRs have not been analyzed in this study; these SNPs are likely to affect the level of gene expression; the impact of SNPs at the 5′-UTRs can be predicted by using some of the RNA assessment tools, such as PreTIS. 81
This study is the first in silico analysis while all other previous studies were next-generation sequencing (NGS) analysis, in vitro analysis, and in vivo analysis14,71,82,83; also, it is the first computational analysis, which revealed that 5 SNPs were identified as highly deleterious in the coding region, whereas 11 SNPs were detected to be damaging in the 3′-UTR, and therefore, may be used as diagnostic markers for AML and might create an ideal target for cancer therapy. These outcomes in combination with all earlier discoveries make AML a model for understanding the philosophies of cancer development.84,85 Finally, clinical techniques are recommended to support these findings.
Conclusions
In this study, the impact of functional mutations in the CEBPA gene was investigated through different bioinformatics analysis techniques, which determined that R339W, R288P, N292S, N292T, and D63N are pathogenic mutations, which have a possible functional influence, and therefore, can be used as diagnostic markers and may assist in genetic studies with a special consideration of the large heterogeneity of AML among different populations. In addition, this study draws attention to 11 SNPs that were identified to be deleterious in the 3′-UTR.
Footnotes
Acknowledgements
The authors wish to acknowledge the enthusiastic cooperation of Africa City of Technology, Sudan.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interest:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MIM and ZOM helped in data curation, methodology, also conceptualized the data, formal analysed the data, illustrated, validated and wrote the manuscript and also helped in drafting the original manuscript. NSM, NME, and AHA helped in data curation, methodology of the data, and also formal analysed the data. MAH conceptualized and validated the manuscript, helped in reviewing & editing, project administration of the data, and also supervised the manuscript.
Data Availability
All data underlying the results are available as part of the article, and no additional source data are required.