
Abstract
Cisplatin resistance in colorectal cancer largely results from the colorectal cancer stem cells which could be targeted to improve the efficacy of chemotherapy. MicroRNAs are possible modulators of cancer stem cell characteristics and maybe involved in the retention of cancer stem cell chemoresistance. The aim of this study was to investigate the biological function of miR-199a/b on cisplatin resistance in colorectal cancer stem cells and its related mechanisms. Here, ALDHA1+ cells from primary colorectal cancer tissues behaved similar to cancer stem cells and were chemoresistant to cisplatin. The presence of a variable fraction of ALDHA1 was detected in 9 out of 10 colorectal cancer specimens. Significantly, increased miR-199a/b expression was detected in ALDHA1+ colorectal cancer stem cells, accompanied by a downregulation of Gsk3β and an overexpression of β-catenin and ABCG2. In patient cohort, enhanced miR-199a/b expression in colorectal cancer tissues was associated with cisplatin response and poor patient survival. In addition, 80% of colorectal cancer samples showed lower level of Gsk3β than their adjacent normal counterparts. Furthermore, Gsk3β was the direct target of miR-199a/b. MiR-199a/b regulated Wnt/β-catenin pathway by targeting Gsk3β in ALDHA1+ colorectal cancer stem cells. By blocking Wnt/β-catenin pathway, we implied that ABCG2 lies downstream of Wnt/β-catenin pathway. ABCG2 was further demonstrated to contribute cisplatin resistance in ALDHA1+ colorectal cancer stem cells and can be regulated by miR-199a/b. Thus, our data suggested that upregulation of miR-199a/b in ALDHA1+ colorectal cancer stem cells contributed to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling, which sheds new light on understanding the mechanism of cisplatin resistance in colorectal cancer stem cells and facilitates the development of potential therapeutics against colorectal cancer.
Introduction
Colorectal cancer (CRC) is one of the most fatal neoplastic diseases worldwide. Although improved treatment strategies have increased the overall survival rates in the early stages, 40%–50% of all CRC patients present with metastasis either at the diagnosis or as recurrent disease upon intended curative therapy. 1 Most CRC patients with distant metastasis are not suitable candidates for conventional intervention and exhibit a poor 5-year survival rate of <10%. 2 Although several types of non-surgical treatment, including cisplatin, 5-fluorouracil, capecitabine, and doxorubicin that are used against advanced CRCs, 3 the 5-year survival rates have not raised substantially in the past decades. 4
Cisplatin resistance is the consequence of a multifactorial event which involves a combination of features. 5 There is increasing evidence proving that resistance to CRC therapy is, at least in part, caused by inherent chemoresistance of a subpopulation of cancer cells. 6 This subpopulation of cancer cells, which is also labeled as cancer stem cells (CSCs) or tumor-initiating cells (TICs), shares many properties with stem cells.7,8 CSCs, which are capable of dictating self-renewal, thus redirecting tumor heterogeneity and tumorigenesis, are not only the source of tumors but have also been associated with tumor aggressiveness and metastasis.6,9 Furthermore, CSCs mediate chemoresistance and subsequent tumor recurrence.6,10 The existence of CRC stem cells (CCSCs) may explain the high rate of tumor relapse after standard therapies. Therapeutic targeting of the CCSCs population is a novel strategy to overcome therapeutic resistance and tumor relapse after CRC treatment. Thus, exploring key regulators and the underlying molecular mechanisms associated with chemoresistance of CCSCs should be focused on.
In CRC, some studies reported that EpCAM, CD133, CD166, and CD44 activities presented the properties to generate new heterogeneous tumor spheres in vitro and tumors in vivo.11–14 However, some controversies are still open about the choice of the most appropriate marker to enrich CCSCs. 13 As for ALDH1, Li et al. 15 found that ALDH1A1 played an important role in tumor aggressiveness and prognosis. Nevertheless, the availability of ALDH1A1 to sort CCSCs needs further study.
MicroRNAs (miRNAs), an abundant class of 19–25 nt non-coding RNAs with important regulatory functions in diverse biological processes, have been demonstrated to be ideal biomarkers and therapeutic intervention targets. 16 MiRNAs are involved in carcinogenesis and cancer progression as oncogenes or tumor suppressors to regulate carcinogenesis and cancer development.17,18 While in stem cells, miRNAs can either promote self-renewal or promote differentiation to determine stem cell fates. 19 However, the roles that miRNA play in chemoresistance of CCSCs and its potential associated mechanisms need to be refined. MiR-199 was reported to be distinctly decreased in hepatocellular carcinoma (HCC) when compared to adjacent non-malignant tissues. 20 It suggested that miR-199 possessed anti-tumor activity and could be used as potential circulating biomarkers for HCC.20,21 However, there are no reports about miR-199 expression in CRC, especially in CCSCs.
Here, we aim to investigate the biological function of miR-199a/b on cisplatin resistance in CCSCs and its related mechanisms. Here, ALDHA1+ cells from primary CRC tissues behave similar to CSCs and are chemoresistant to cisplatin. The presence of a variable fraction of ALDHA1 was detected in 9 out of 10 CRC specimens. Significantly, increased miR-199a/b expression is detected in ALDHA1+ CCSCs, accompanied by a downregulation of Gsk3β and an overexpression of β-catenin and ABCG2. In patient cohort, enhanced miR-199a/b expression in CRC tissues is associated with cisplatin response and poor patient survival. In addition, 80% of CRC samples show lower level of Gsk3β than their adjacent normal counterparts. Furthermore, Gsk3β was the direct target of miR-199a/b. MiR-199a/b regulates Wnt/β-catenin pathway by targeting Gsk3β in ALDHA1+ CCSCs. By blocking Wnt/β-catenin pathway, we imply that ABCG2 lies downstream of Wnt/β-catenin pathway. ABCG2 has been further demonstrated to contribute to cisplatin resistance in ALDHA1+ CCSCs and can be regulated by miR-199a/b. Thus, our data suggested that upregulation of miR-199a/b in ALDHA1+ CCSCs contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling.
Materials and methods
Animal and cell culture
Female athymic BALB/c nu/nu mice, 3–4 week-old, obtained from HFK Bioscience (China), were maintained at the Animal Core Facility at The First Affiliated Hospital of Xinxiang Medical University, under specific pathogen-free (SPF) condition. All studies on mice were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and were approved by the ethical committee of The First Affiliated Hospital of Xinxiang Medical University.
Sample collection
Overall, 10 patients with primary CRC, who consecutively underwent chemotherapy at Department of Gastroenterology, The First Affiliated Hospital of Xinxiang Medical University, were enrolled into this study from January 2009 to June 2015. Informed consent for the additional core needle biopsy and experimental use of tumor samples was obtained from all patients, following a protocol approved by the Ethics Committee of The First Affiliated Hospital of Xinxiang Medical University. The tumor response was evaluated by the Response Evaluation Criteria in Solid Tumors (RECIST), which was defined in Supplemental Information.
Flow cytometry assay and fluorescence activated cell sorting
Flow cytometry assay was done on single-cell suspensions obtained by enzymatic digestion of spheres which were derived from primary CRC samples and labeled with Alexa fluor@ 488 conjugated anti-ALDHA1 antibody (Abcam, USA) by using an Epics Altra flow cytometer (Beckman Coulter, USA).
Fluorescence activated cell sorting (FACS): Spheres which were derived from primary tumor samples were dissociated into single cells by enzymatic digestion. Single-cell suspensions were washed and incubated with staining solution 1% bovine serum albumin (BSA) and 2mM ethylenediaminetetraacetic acid (EDTA) with the specific antibodies at appropriate dilutions. The details are listed in the Supplemental Information.
Sphere formation and propagation
Single-cell suspensions were obtained from CRC primary tissue samples. CRC primary tissue samples were shipped to laboratory in cold RPMI-1640 medium with penicillin/streptomycin within 1 h of removal from patients. Surgical specimens were washed with cold phosphate-buffered saline (PBS) supplemented with high doses of penicillin/streptomycin three times, chopped with a sterile blade, and incubated with 1 mg/mL collagenase II (Sigma-Aldrich, USA) for 30 min at 37°C. The details are listed in the Supplemental Information.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
ALDHA1+ cells, ALDHA1− cells, and differentiated adherent progeny of ALDHA1+ cells were seeded on 96-well plates at 2000 cells/well. Cells were then treated with increasing concentrations of doxorubicin from 0 to 20 µg/mL for 24 h. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich) was used to determine relative cell growth every 24 h for cell growth curves, which was defined in Supplemental Information.
Quantitative real-time reverse transcription polymerase chain reaction analysis
Total RNA was extracted using TRIzol (Invitrogen, USA) and treated with RNase-free DNase (QIAGEN, USA). Mature miRNA expression analysis was conducted using a TaqMan MicroRNA Assays (Applied Biosystems, USA). Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR; qPCR) analysis was performed using a SYBR Green Reagents (Bio-Rad, USA) on the iQ5 Real-Time PCR Detection System (Bio-Rad). The relative expression levels of the miRNAs in tissue and cells were normalized to U6 and those in the serum and exosomes were normalized to the insert control according to the manufacturer’s protocol.
MiRNA luciferase assay
A pMIR-REPORT™ luciferase reporter vector with a miR-199a/b target sequence cloned into its 3′ untranslated region (3′-UTR) (Invitrogen) was used. Luciferase activity was assayed using a Luciferase Assay Kit (Promega, USA).
Lentiviral construction and infection
For overexpression of miR-199a/b, the pcDNATM6.2-GW/EmGFP-miR vector was recombined into pLenti6/V5 using LR clonase (Invitrogen), resulting in the generation of two pLenti6/V5-miR-199a/b plasmids. Cells were transfected with viral supernatant, as described earlier. 22
MiR-199a/b-depleted by lentiviral infection
To inhibit endogenous miR-199a/b, cells were transfected with the antagomir-199a/b lentivector (System. BioSciences (SBI), USA) or negative control (anti-nc lentivector). 23 Levels of antagomir-199a/b lentivirus expressing anti-miR-199a/b were determined using the synthetic miR-199a/b binding site–containing luciferase reporter gene. Western blotting is detailed in Supplemental Information.
Xenograft assay
Female athymic BALB/c nu/nu mice at 3–4 weeks of age were injected subcutaneously into the right flank with 1 × 104 of ALDHA1+ CCSCs and 1 × 104 of ALDHA1− cells. The xenografts were monitored until the tumor volumes reached ~100 mm3; each group was randomized into two subgroups that were either left untreated or received intraperitoneal injections of doxorubicin (4 mg/kg) every 5 days (three cycles). The tumor volume was measured with a caliper using the following formula: V (mm3) = 0.52 × length (mm) × width2 (mm2).
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed by the DeadEnd™ Fluorometric TUNEL system according to the manufacturer’s instructions (Promega) in xenograph tumor tissues derived from lenti-si-ABCG2-CCSCs, lenti-si-NC-CCSCs group, and untransduced ALDHA1+ CCSCs following treatment with or without three cycles of cisplatin. Cells were then observed under a fluorescence microscope (Olympus 200 Optical Co. Ltd, Germany), and a nucleus with bright green fluorescence staining was recorded as a TUNEL-positive event.
Statistical analysis
SPSS13.0 software was used. Each experiment was performed at least three times. The data were expressed as mean ± standard deviation (SD) and one-way analysis of variance (ANOVA). An unpaired Student’s t test was used to determine the significant differences of all the results. Significances are, ***p < 0.001; **p < 0.01; and *p < 0.05.
Results
ALDHA1+ spheres from primary CRC tissue samples display stem cell–like features
To identify whether ALDHA1 is the surface marker to sort CCSCs, we analyzed 10 fresh tumor samples derived from a series of CRC patients. Flow cytometry demonstrated the presence of a variable fraction of ALDHA1, ranging from 0.1% to 9.7%, in 9 out of 10 CRC specimens. CSCs are believed to be able to form spheres in serum-free cultivation. Therefore, we cultured two cases of primary CRC tissue samples (Table S1) to induce sphere formation in serum-free cultivation. After culturing for 2–4 weeks, although a large number of cells died, some tumor cells grew to form spheres (data were not shown). The tumor spheres were at least passaged 10 times, indicating the self-renewal of these tumor sphere cells. Because the definition of CSCs relies mainly on stem cell–like properties, we further fractionated ALDHA1+ and ALDHA1− cells derived from clinical case CC051 and CC126 by FACS to corroborate the capacity of self-renewal, differentiation, and tumorigenesis in ALDHA1+ spheres. ALDHA1+ cells could form compact self-renewing spheres whereas ALDHA1− cells could not (Figure 1(a)). Significantly, with increasing concentrations of cisplatin from 0.5 to 20 µg/mL, cell viability in both ALDHA1− cells and differentiated adherent progeny of ALDHA1+ cells decreased at each concentration compared with that in ALDHA1+ cells (Figure 1(b)). In addition, we found that the expression of ALDHA1 decreased gradually when ALDHA1+ spheres were cultured in a serum-containing medium (Figure S1). To evaluate the tumorigenic potential of ALDHA1+ cells, the same numbers of ALDHA1+ and ALDHA1− cells (104 cells) were subcutaneously injected into the flank of nude mice. We found that tumor formation of ALDHA1+ cells was faster and resulted in increased tumor take compared with that observed in ALDHA1− cells (Figure 1(c)). To investigate whether CRCs originating from ALDHA1+ cells possess higher long-term tumorigenic potential compared with the rarely observed tumors originating from the ALDHA1− cells, we performed serial transplantation assays in nude mice of cells isolated from CC051 or CC126 tumor xenografts originally derived from ALDHA1+ or ALDHA1− cells injection. Cells derived from ALDHA1+ tumors were able to generate tumors in primary, secondary, and tertiary transplantation, whereas cells from ALDHA1− tumors lost tumorigenic potential during serial transplantations (Figure 1(c)). Therefore, these data indicated that ALDHA1+ cells from CRC patient–derived tumor spheres bear stem cell–like features.
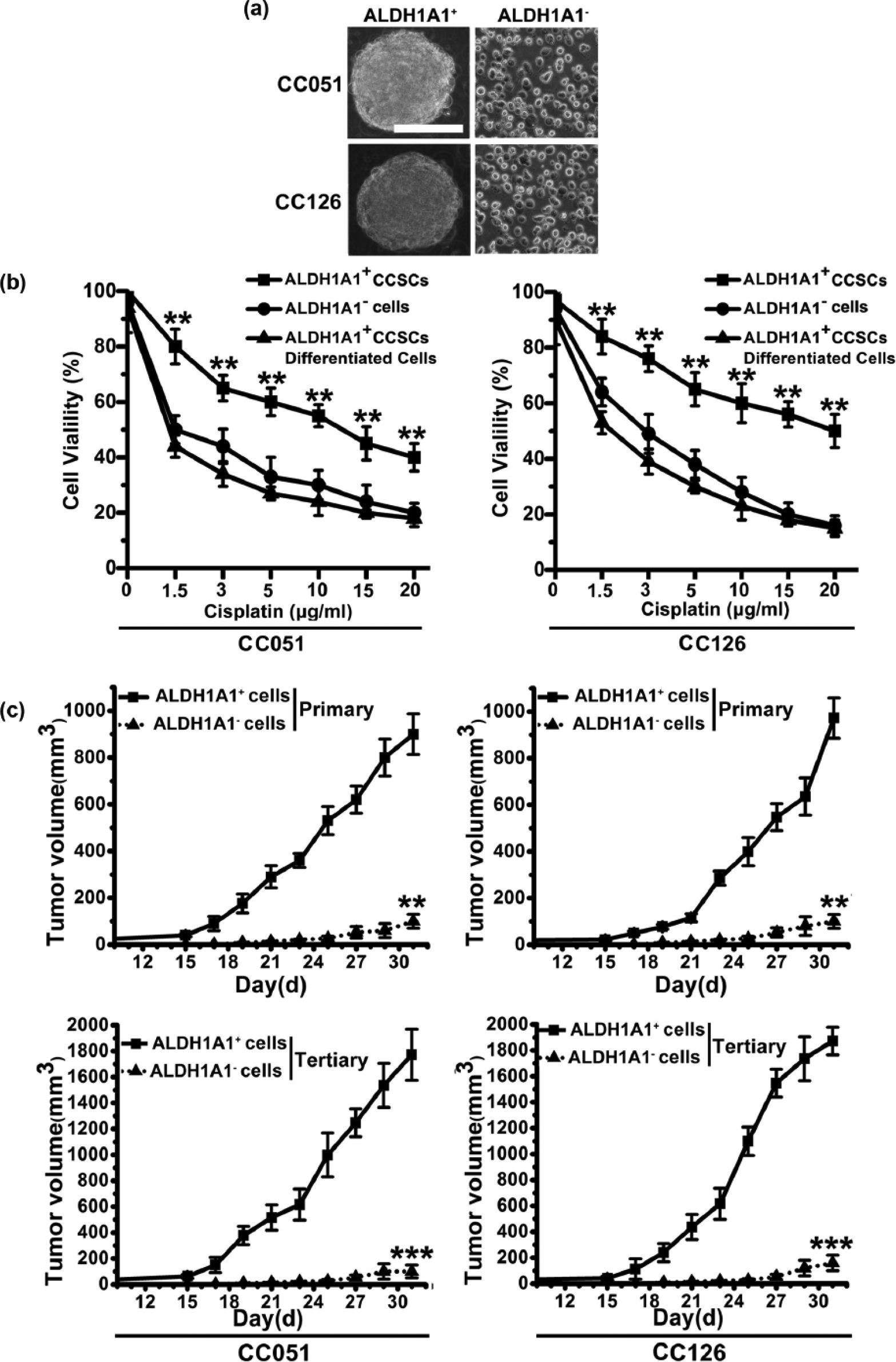
ALDHA1+ spheres from primary CRC tissue samples display stem cell–like features. (a) Phase-contrast images of ALDHA1+ sphere cells and ALDHA1− cells (scale bar: 100 µm). (b) With the presence of cisplatin, cell viability of ALDHA1+ sphere cells, ALDHA1− cells, and differentiated adherent progeny of ALDHA1+ spheres were determined by MTT (columns: mean of three individual experiments; SD; **p < 0.01). (c) In vivo serial transplantation assay. A total of 104 ALDHA1+ cells and ALDHA1− cells, purified from clinical case CC051 and CC126, were injected subcutaneously into nude mice. Derived tumor xenografts were dissociated into single-cell suspension and then serially re-injected in mice (104 cells), generating secondary and then tertiary tumors. Tumor growth curves of primary and tertiary tumors are shown (columns: mean of three individual experiments; SD; ***p < 0.001; **p < 0.01).
MiR-199a/b expression is enhanced in ALDHA1+ CCSCs
To obtain the expression profile of miR-199a/b in ALDHA1+ CCSCs, we compared miR-199a/b expression in ALDHA1+ CCSCs with that in differentiated adherent progeny of ALDHA1+ CCSCs and ALDHA1− cells. We found that the expression of miR-199a/b in ALDHA1+ CCSCs was significantly increased at messenger RNA (mRNA) level compared with that in differentiated adherent progeny of ALDHA1+ CCSCs and ALDHA1− cells (Figure 2(a)). To further explore the targeting function of miR-199a/b, we evaluated the percentage of luciferase suppression by transduction of ALDHA1+ CCSCs with miR-199a/b antagomir and negative control (anti-nc). We found that luciferase activity of ALDHA1+ CCSCs (un) and ALDHA1+ CCSCs transduced with anti-nc was suppressed (Figure 2(b)). The suppression of luciferase activity in ALDHA1+ CCSCs transduced with antagomir-199a/b was significantly restored (Figure 2(b)). Thus, these findings indicate that the expression as well as the target function of miR-199a/b is enhanced in ALDHA1+ CCSCs.

MiR-199a/b expression is increased in ALDHA1+ CCSCs. (a) Relative expression of miR-199a/b in ALDHA1+ CCSCs, differentiated adherent progeny of ALDHA1+ CCSCs, and ALDHA1− cells were examined by qPCR (columns: mean of three individual experiments; SD; **p < 0.01 vs untransduced cells). (b) The percentage of luciferase suppression was measured in ALDHA1+ cells and ALDHA1+ CCSCs including ALDHA1+ CCSCs without treatment (un) and ALDHA1+ CCSCs transduced with miR-199a/b antagomir and negative control (anti-nc; columns: mean of three individual experiments; SD; **p < 0.01).
Upregulation of miR-199a/b expression contributes to cisplatin resistance in ALDHA1+ CCSCs
To investigate whether the upregulation of miR-199a/b expression in ALDHA1+ CCSCs contributes to cisplatin resistance, in vivo assay was taken. We found that xenograft tumors in antagomir-199a/b-CCSCs group were slightly smaller than that in anti-nc-CCSCs group and untransduced group (Figure 3(a)), although no significant difference was observed. Upon intratumor injection with cisplatin, the size of xenograft tumors in antagomir antagomir-199a/b-CCSCs group was significantly reduced in comparison with that in anti-nc-CCSCs group and untransduced group (Figure 3(a)). We, next, detected cisplatin-induced apoptosis by using Annexin V/propidium iodide (PI) staining in antagomir-199a/b-CCSCs, anti-nc-CCSCs, and untransduced CCSCs. In the presence of cisplatin, antagomir-199a/b-CCSCs showed increased percentage of Annexin V+PI+/Annexin V+PI−cells about 5- to 14- fold as compared with anti-nc-CCSCs and untransduced CCSCs (Figure 3(b)). However, in the absence of cisplatin, antagomir-199a/b-CCSCs showed increased percentage of apoptotic and necrotic cells about only 1- to 4- fold as compared with anti-nc-CCSCs and untransduced CCSCs (Figure 3(b)). Our data demonstrate that declined miR-199a/b expression by antagomir sensitizes ALDHA1+ CCSCs to cisplatin via apoptosis.

Reduced miR-199a/b expression by antagomir-199a/b sensitizes ALDHA1+ CCSCs to cisplatin via apoptosis. (a) The potential of tumor initiation of untransduced ALDHA1+ CCSCs, antagomir-199a/b-CCSCs, or anti-nc-CCSCs fractions were tested by subcutaneous injection, and representative tumor volumes were measured following treatment with or without three cycles of cisplatin (columns: mean of three individual experiments; SD; **p < 0.01). (b) With or without cisplatin, the percentage of Annexin V+PI+/Annexin V+PI−cells was measured by flow cytometry.
MiR-199a/b targets Gsk3β directly
To explore the molecular mechanisms of miR-199a/b action on resistance to cisplatin in ALDHA1+ CCSCs, we integrated mRNA expression profiling findings with prediction algorithms of TargetScan, a miRBase algorithm. We found that the seed sequence of miR-199a/b had a perfect match with the 3′-UTR of the Gsk3β mRNA (Figure 4(a)). To validate whether Gsk3β is a direct target of miR-199a/b, wild sequence of Gsk3β 3′-UTR and its corresponding mutant counterparts were cloned into pMIR-REPORT luciferase vector. Reporter activities in wild type 3′-UTR (WT) were promoted by miR-199a/b antagomir but not in mutant 3′-UTR (MT) (Figure 4(b)). These results indicated that miR-199a/b downregulated Gsk3β expression by directly targeting its 3′-UTR. To further examine whether miR-199a/b affects Gsk3β expression, we transduced ALDHA1+ CCSCs with lenti-miR-199a/b or antagomir-199a/b. Western blot analysis revealed that miR-199a/b overexpression triggered a repressive effect on endogenous Gsk3β but stimulated β-catenin protein expression (Figure 4(c)). Conversely, in miR-199a/b depleted cells (antagomir-199a/b-CCSCs), expression of Gsk3β was increased whereas β-catenin expression was suppressive (Figure 4(c)). Therefore, our data indicated that miR-199a/b regulated Gsk3β expression by directly targeting their 3′-UTRs. The increase of miR-199a/b might result in the reduction of Gsk3β protein by relieving translational repression at the 3′-UTRs of Gsk3β.
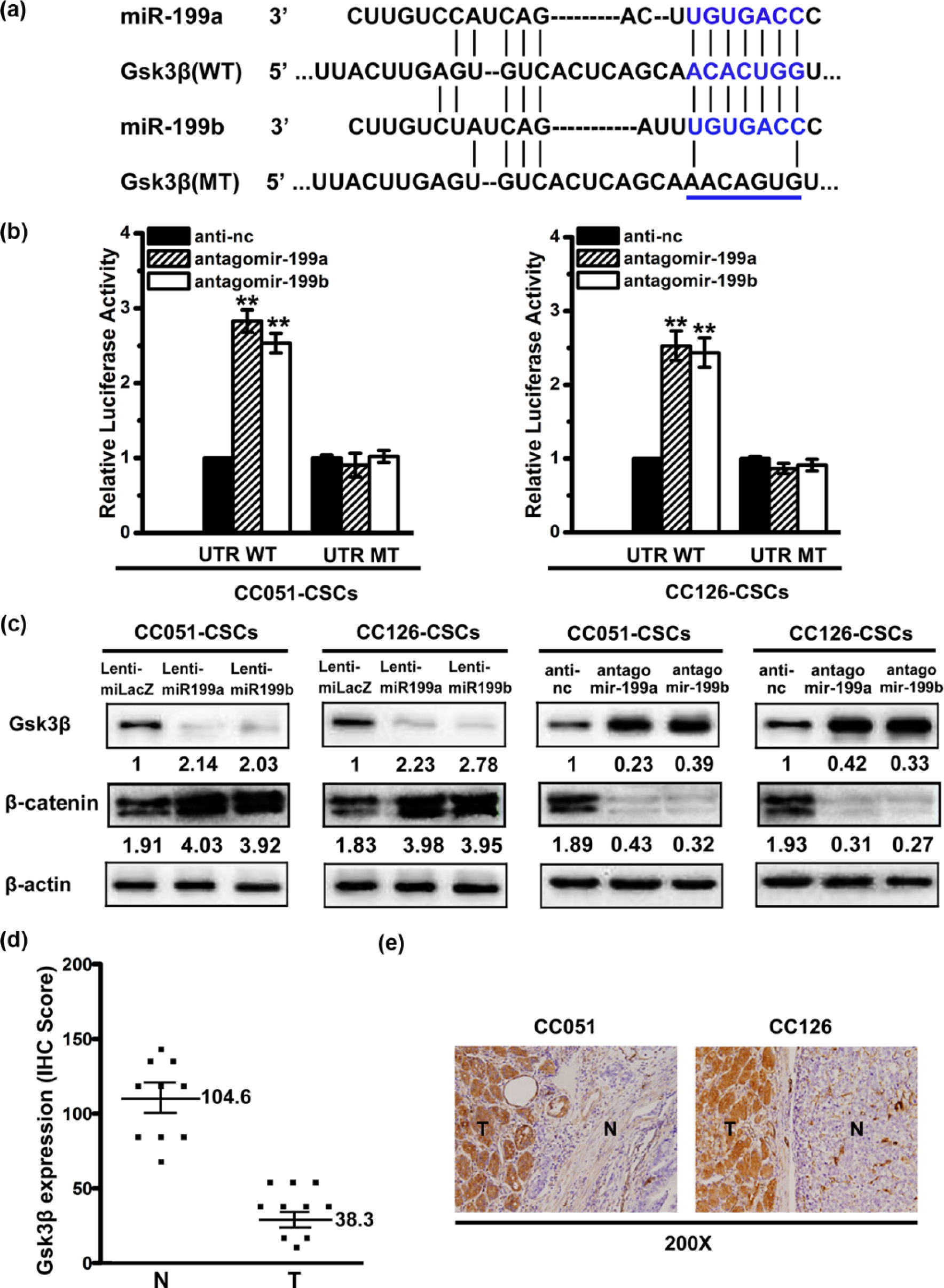
MiR-199a/b targets Gsk3β. (a) Alignment of the putative miR-199a/b binding site to Gsk3β 3′-UTR. The seed region of wild type and mutant 3′-UTR of Gsk3β, shown with underlines and base substitutions in blue, were cloned for the luciferase reporter assay. (b) Wild-type and mutant 3′-UTRs of Gsk3β were cloned, and the luciferase reporter assay was performed. (c) Gsk3β and β-catenin expression was examined via western blotting after miR-199a/b knockdown in ALDHA1+ CCSCs. (d) Expression of Gsk3β was determined based on the IHC score in both CRC tumor samples and adjacent normal tissues. Mann–Whitney U test was used for comparisons between the two groups. (e) β-catenin was clearly upregulated in tumor tissues.
To establish clinicopathological associations, Gsk3β expression patterns in CRC and adjacent normal tissues were examined with immunohistochemistry (IHC). The mean IHC scores of tumor samples versus adjacent normal tissues were 38.3 and 104.6, with significant differences (p < 0.01; Figure 4(d)). Moreover, 80% of CRC tumor samples showed higher levels of β-catenin than their adjacent normal counterparts (Figure 4(e)).
Upregulation of miR-199a/b in ALDHA1+ CCSCs contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling
The expression of adenosine triphosphate–binding cassette (ABC) transporters, which are putative to mediate the inherent drug efflux capability, including ABCG1, ABCA1, ABCG2, ABCA2, ABCC1, and ABCC2, was detected in antagomir-199a/b-CCSCs compared with that in anti-nc-CCSCs and untransduced CCSCs. We found that the expression of ABCG1, ABCA1, and ABCG2 was decreased in antagomir-199a/b-CCSCs compared with that in anti-nc-CCSCs and untransduced CCSCs by using qPCR (Figure 5(a)). To further characterize the association between Wnt/β-catenin pathway and ABC transporters, we used XAV-939 (the inhibitor of Wnt/β-catenin pathway) to block the activity of Wnt/β-catenin pathway. We found that the expression of β-catenin and ABCG2 was significantly decreased in the presence of XAV-939 (Figure 5(b)), which suggested that Wnt/β-catenin pathway was one of the upstream mechanisms to regulate the expression of ABCG2 in ALDHA1+ CCSCs. However, the expression of ABCG1 and ABCA1 was almost unchanged at protein level with and without the presence of XAV-939 (Figure 5(b)).

MiR-199a/b contributes to cisplatin resistance through Wnt/β-catenin-ABCG2 signaling. (a) Relative expression of ABCG1, ABCA1, ABCG2, ABCA2, ABCC1, and ABCC2 in antagomir-199a/b-CCSCs, anti-nc-CCSCs, and untransduced CCSCs were examined by qPCR (columns: mean of three individual experiments; SD; **p < 0.01 vs untransduced cells). (b) Western blot analysis was performed for β-catenin, ABCG1, ABCA1, and ABCG2 in ALDHA1+ CCSCs with or without XAV-939. (c) The population of viable cells of ALDHA1+ CCSCs upon transduction with antagomir-199a/b, anti-nc, lenti-si-ABCG2, or lenti-si-nc was measured by MTT when they were all treated with increasing concentrations of cisplatin (columns: mean of three individual experiments; SD; **p < 0.01).
To further investigate the link between the cisplatin resistance and the expression of miR-199a/b and ABCG2, a lentiviral-based approach was used to knockdown ABCG2 with lenti-si-ABCG2. Without cisplatin, the cell viability of ALDHA1+ CCSCs was not significantly changed upon transduction with antagomir-199a/b, anti-nc, or lenti-si-ABCG2 (Figure 5(c)), suggesting that the upregulation of miR-199a/b and ABCG2 were not required for ALDHA1+ CCSCs survival. With increasing concentration of cisplatin, the percentage of viable cells in antagomir-199a/b-CCSCs and lenti-si-ABCG2-CCSCs decreased more rapidly than that in anti-nc-CCSCs and transduced CCSCs (Figure 5(c)). At each concentration, cisplatin induced the highest growth inhibition in ALDHA1+ CCSCs with the transduction of lenti-si-ABCG2 (Figure 5(c)).
Moreover, we built xenograft tumor model by using ALDHA1+ CCSCs, lenti-si-nc-CCSCs, or lenti-si-ABCG2-CCSCs. We found that xenograft tumors in lenti-si-ABCG2-CCSCs group were slightly smaller (Figure 6(a)), although no significant difference was observed. Upon intratumor injection with cisplatin, the size of xenograft tumors in lenti-si-ABCG2-CCSCs group was significantly reduced in comparison with that in lenti-si-nc-CCSCs and untransduced ALDHA1+ CCSCs (Figure 6(a)). In addition, to detect whether cisplatin-mediated cell death is generated by apoptosis, TUNEL apoptosis index was examined in every tumor group. Intratumor injection with cisplatin in xenograft tumors derived from ALDHA1+ CCSCs resulted in an increased TUNEL index compared with intratumor injection with glucose (control) (Figure 6(b)). Significantly, with the treatment of cisplatin, xenograft tumors in lenti-si-ABCG2-CCSCs group showed an obviously higher TUNEL index than those in lenti-si-NC-CCSCs group and untransduced ALDHA1+ CCSCs (Figure 6(b)). Taken together, our data suggested that upregulation of miR-199a/b in ALDHA1+ CCSCs contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling.

High ABCG2 expression promotes cisplatin resistance in ALDHA1+ CCSCs via apoptosis in vivo. (a) The potential of tumor initiation of ALDHA1+ CCSCs (untransduced), lenti-si-ABCG2-GCSCs, and lenti-si-nc-GCSCs fractions was tested by subcutaneous injection, and representative tumor volumes were measured following treatment with or without three cycles of cisplatin (columns: mean of three individual experiments; SD; **p < 0.01). (b) Apoptosis in xenograft tumors derived from ALDHA1+ CCSCs (untransduced), lenti-si-ABCG2-GCSCs, and lenti-si-NC-GCSCs following treatment with or without three cycles of cisplatin was detected by TUNEL assay (scale bar: 100 µm; columns: mean of three individual experiments; SD; **p < 0.01).
To test the hypothesis, we characterized the associations among miR-199a/b, Wnt/β-catenin pathway, and ABCG2. We found that the expression of β-catenin and ABCG2 were reduced, while Gsk3β was upregulated in ALDHA1+ CCSCs transduced with antagomir-199a/b (Figure 7(a)). With the presence of XAV-939, the expression of β-catenin and ABCG2 in ALDHA1+ CCSCs was downregulated (Figure 7(a)). The expression of ABCG2 was reduced in ALDHA1+ CCSCs transduced with lenti-si-ABCG2, whereas the expression of Gsk3β and β-catenin was unchanged compared with those in untreated ALDHA1+ CCSCs (Figure 7(a)). Significantly, with the presence of XAV-939, the expression of ABCG2 and β-catenin in ALDHA1+ CCSCs transduced with lenti-miR-199a/b restored compared with those in untransduced ALDHA1+ CCSCs with the presence of XAV-939 (Figure 7(a)).

The associations among miR-199a/b, Wnt/β-catenin pathway, and ABCG2, and the correlation between miR-199a/b and chemoresistance in patient cohort are defined. (a) Protein level of miR-199a/b, Gsk3β, β-catenin, and ABCG2 was detected in ALDHA1+ CCSCs upon transduction with lenti-miR-199a/b, lenti-si-ABCG2, or antagomir-199a/b alone or combined. (b) The correlation of clinical response and miR-199a/b expression in 10 CRC patients (CR: disappearance of the disease; PR: reduction of >30%; PD: enlargement >20%) was built. (c) Kaplan–Meier survival curves for colorectal cancer patients plotted against miR-199a/b expression showed a statistical difference in survival between patients with high and low miR-199a/b.
Upregulation of miR-199a/b in CRC tissues correlates chemotherapy resistance and poor patient survival
To investigate whether miR-199a/b expression is related to chemoresistance in CRC patient cohort, 10 CRC patients who exhibited CR (chemosensitive), PR (chemosensitive), or PD (chemoresistant) according to RECIST criteria were enrolled. We found that miR-199a/b expression in PD tissues was the highest among the different chemoresponsive CRC tissues (Figure 7(b)). It implies a link between miR-199a/b expression and tumor response in CRC patients. Survival curve showed that patients with low miR-199a/b expression survived obviously longer than patients with low miR-199a/b expression (Figure 7(c)).
Discussion
CSCs display enhanced self-renewal ability and high expression of stem cell markers, both of which have been used as surrogate markers for frequently correlated characteristics that are arguably more important clinically: chemoresistance and tumorigenicity.24,25 However, the molecular base of chemoresistance in CSCs to conventional therapies remains incompletely understood.
Studies of CSCs rely on experimental isolation from bulk tumor cells, which is most frequently based on expression of stem cell markers such as CD133 26 and CD44. 27 Some controversies, however, are still open about the choice of the most appropriate marker to enrich for CCSCs.11,13 As for ALDH1, Li et al. 15 found that ALDH1A1 played an important role in tumor aggressiveness and prognosis. Nevertheless, the availability of ALDH1A1 to sort CCSCs needs further study. Here, we analyzed 10 fresh tumor samples derived from CRC patients and found the presence of a variable fraction of ALDHA1 in 9 out of 10 CRC specimens. We found that the ability of sphere formation, self-renewal, chemoresistance to cisplatin, and initiating tumors in vivo in ALDHA1+ cells were enhanced compared with ALDHA1− cells. In addition, we found that the expression of ALDHA1 decreased gradually when ALDHA1+ spheres were cultured in a serum-containing medium.
MiR-199 was reported to be distinctly decreased in HCC when compared to adjacent non-malignant tissues,20,28 but the exact role and associated mechanisms of miR-199 in HCC were blurred.20,21 In CRC, especially in CCSC, there are no reports about its expression. Moreover, the role and associated mechanisms are unclear. Here, we found that the expression as well as the target function of miR-199a/b was increased in ALDHA1+ CCSCs. Our data demonstrated that downregulation of miR-199a/b expression by antagomir sensitized ALDHA1+ CCSCs to cisplatin via apoptosis. Then, we found that the seed sequence of miR-199a/b had a perfect match with the 3′-UTR of the Gsk3β mRNA. In addition, western blot analysis clearly suggested that miR-199a/b could regulate Wnt/β-catenin pathway by directly targeting Gsk3β in ALDHA1+ CCSCs. In patient cohort, the mean IHC scores of Gsk3β in CRC tumor samples were found to be decreased compared with adjacent normal tissues.
A number of molecular mechanisms about chemoresistance have been proposed, including the upregulation of multidrug transporters from the ABC superfamily and resistance to apoptosis. 29 ABCG2 is a major multidrug resistance pump that renders cancer cells resistance to chemotherapeutic drugs. 30 The increased expression of ABCG2 has been linked to multidrug resistance (MDR) in cancer, and there have also been numerous studies describing ABCG2-mediated transport of chemotherapeutic drugs including mitoxantrone, methotrexate, topotecans, and cisplatin. 31 In this study, our data suggested that upregulation miR-199a/b expression directly contributed to their cisplatin resistance by regulating ABCG2 through Wnt/β-catenin pathway. Then, we characterized the associations among miR-199a/b, Wnt/β-catenin pathway, and ABCG2 to test the hypothesis. In addition, we found that miR-199a/b expression in PD CRC tissues was the highest among the different chemoresponsive CRC tissues. Survival curve showed that patients with low miR-199a/b expression survived obviously longer than patients with low miR-199a/b expression. These data suggested a potential predicted value of miR-199a/b on chemoresistance in patient cohort.
In conclusion, we found that ALDHA1+ cells from primary CRC tissues behave similar to CSCs and are chemoresistant to cisplatin. The presence of a variable fraction of ALDHA1 was detected in 9 out of 10 CRC specimens. Significantly, increased miR-199a/b expression is detected in ALDHA1+ CCSCs, accompanied by a downregulation of Gsk3β and an overexpression of β-catenin and ABCG2. In patient cohort, enhanced miR-199a/b expression in CRC tissues is associated with cisplatin response and poor patient survival. In addition, 80% of CRC samples showed lower level of Gsk3β than their adjacent normal counterparts. Furthermore, Gsk3β was the direct target of miR-199a/b. MiR-199a/b regulates Wnt/β-catenin pathway by targeting Gsk3β in ALDHA1+ CCSCs. By blocking Wnt/β-catenin pathway, we imply that ABCG2 lies downstream of Wnt/β-catenin pathway. ABCG2 is further demonstrated to contribute to cisplatin resistance in ALDHA1+ CCSCs and can be regulated by miR-199a/b. Thus, our data suggested that upregulation of miR-199a/b in ALDHA1+ CCSCs contributes to cisplatin resistance via Wnt/β-catenin-ABCG2 signaling.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.