
Abstract
Keloid is a disorder of fibroproliferative diseases that occurs in wounds, characterized by an exaggerated response to injury. The key factor responsible for the disease process has not been identified. This study sought to elucidate the role of β-catenin in the regulation of keloid phenotypes and signaling. Expression of β-catenin in keloid and normal non-keloid samples was measured by real-time polymerase chain reaction. Knockdown of β-catenin was achieved by delivering small interfering RNA to target β-catenin. Cell proliferation, cell cycle progression, and apoptosis of keloid cells were measured by functional assays in vitro. The proteins related to keloid fibrosis were measured by Western blotting. β-catenin expression was significantly upregulated in keloid tissue samples compared with the normal non-keloid age-adjusted skin sample counterparts. Functionally, targeting β-catenin with lipofection-delivered small interfering RNA oligonucleotide inhibited the proliferation and cell cycle arrest in G0/G1 phase and increased apoptosis of fibroblast cells, accompanied by downregulation of Wnt2 and cyclin D1 as well as the phosphorylation level of glycogen synthase kinase 3 beta in the keloid fibrosis. Our study supports a crucial role of β-catenin in the regulation of fibroproliferation and extracellular matrix deposition. Targeting β-catenin using small interfering RNA oligonucleotide may be a promising approach for preventing excessive fibroproliferative development after wound healing and may lead to the development of novel strategies for restoring keloid diseases.
Introduction
Keloid is a fibroproliferative disorder that occurs in wounds as an exaggerated response to injury and is characterized by overproduction of extracellular matrix (ECM). Keloids do not regress spontaneously, and they are resistant to treatment and tend to recur after excision. 1 Relative to other chronic skin disease, keloid causes disfigurement or disability disease healing and significantly affects the quality of life. 2 The key factor responsible for the disease process has not been identified and thus, there is no satisfactory treatment. 3
Some researchers reported differential patterns of gene expression in the synthesis of ECM induced by several regulators of wound healing. For example, growth factors have been implicated in the pathogenesis of keloids, including transforming growth factor beta, epidermal growth factor, fibroblast growth factors, and platelet-derived growth factor,4,5 and resistance of keloid fibroblasts to hydrocortisone (HC) downregulation of connective tissue growth factor, insulin-like growth factor (IGF)-binding protein 3, elastin, and types I, III, and V collagen gene expression.6,7 However, the molecular mechanisms underlying keloid formation are unclear. Recent reports about self-renewal or repair have focused on the Wnt/β-catenin signaling pathway;8–10 there have been also no precise studies of Wnt/β-catenin signaling pathway in keloid fibroblasts.
In this study, we show that β-catenin was remarkably upregulated in human keloid fibroblast tissues and their isolated fibroblasts as compared with the normal counterparts. We also demonstrate that targeting β-catenin with small interfering RNA (siRNA) resulted in sustained inhibition of profibrotic phenotypes and suppression of β-catenin, Wnt2, glycogen synthase kinase 3 beta (GSK3β), and cyclin D1 expression. Our findings suggest that downregulation of β-catenin in keloid fibroblast tissues could be an effective approach to therapeutic method.
Materials and methods
The experimental specimens and cell culture
The experimental specimens were derived from plastic surgery department of Second Affiliated Hospital of Fujian Medical College, which were taken from the face, chest, back, abdomen, or limbs. The study protocol was approved by the ethics committee of our institution (Second Affiliated Hospital of Fujian Medical University), and informed consent was obtained from all subjects. Clinically typical, untreated keloid tissue samples were obtained from 62 patients using standard surgical procedures, and normal age-adjusted skin samples were obtained from 21 individuals without keloids. All subjects were Chinese and ranged in age from 2 to 62 years (mean ± standard error (SE): 30.19 ± 14.69 years for keloid patients and 30.76 ± 15.86 years for normal individuals). Specimens were from patients with no skin disease, connective tissue disease, or organ disease and from those who were not on chemotherapy.
Primary fibroblast cultures were made using the explant method. Briefly, explant tissue samples were cut into small pieces that were washed twice in phosphate-buffered saline (PBS), placed in 6-cm culture dishes, and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM (with 2 mM
siRNA preparation and treatment
The three siRNA oligonucleotides (oligos) were selected for human β-catenin gene (GenBank accession: NM_019178) and were purchased from GenePharma (Shanghai, China). The target sense sequences of si-β-catenin RNA were as follows-(1) 5′-CGGAGGAGAUGUACAUUCAdTdT-3′, (2) 5′-GAUGGGAUCAAACCUGACAdTdT-3′, and (3) 5′-GACCCUCUCAGAACCAAAUdTdT-3′. The sequence of negative control was as follows-5′-UUCUCCGAACGUGUCACGUdTdT-3′. The siRNAs were transfected into human keloid fibroblast and normal fibroblast cells using Lipofectamine 2000 reagent (Invitrogen), following the manufacturer’s procedure, at a concentration of 50 nM. The fluorescent labeled siRNA (FAM-siRNA) was used to detect transfection efficiency using a fluorescence microscope.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was isolated from human keloid, normal control tissues, and fibroblast cells using TRIzol reagent (Invitrogen). PrimeScript RT reagent Kit (TaKaRa Bio Inc., Kusatsu, Japan) was used to perform the reverse transcription. For β-catenin, Wnt2, GSK3β, and cyclin D1 expression detection, quantitative real-time polymerase chain reaction (qRT-PCR) was performed using SYBR Premix Ex Taq II (TaKaRa Bio Inc.). The β-actin was used for normalization. Each sample was performed in triplicate and on the ABI PRISM® 7300 System according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). The cycle parameters for the PCR reaction was 95°C for 15 min followed by 40 cycles of a denaturing step at 95°C for 10 s and an annealing/extension step at 60°C for 60 s. The sequences of gene-specific primers are listed in Table 1. All reactions were calculation of gene expression and were based on the comparative CT [2(-Delta Delta C(T))] method. 11
qRT-PCR primers design.
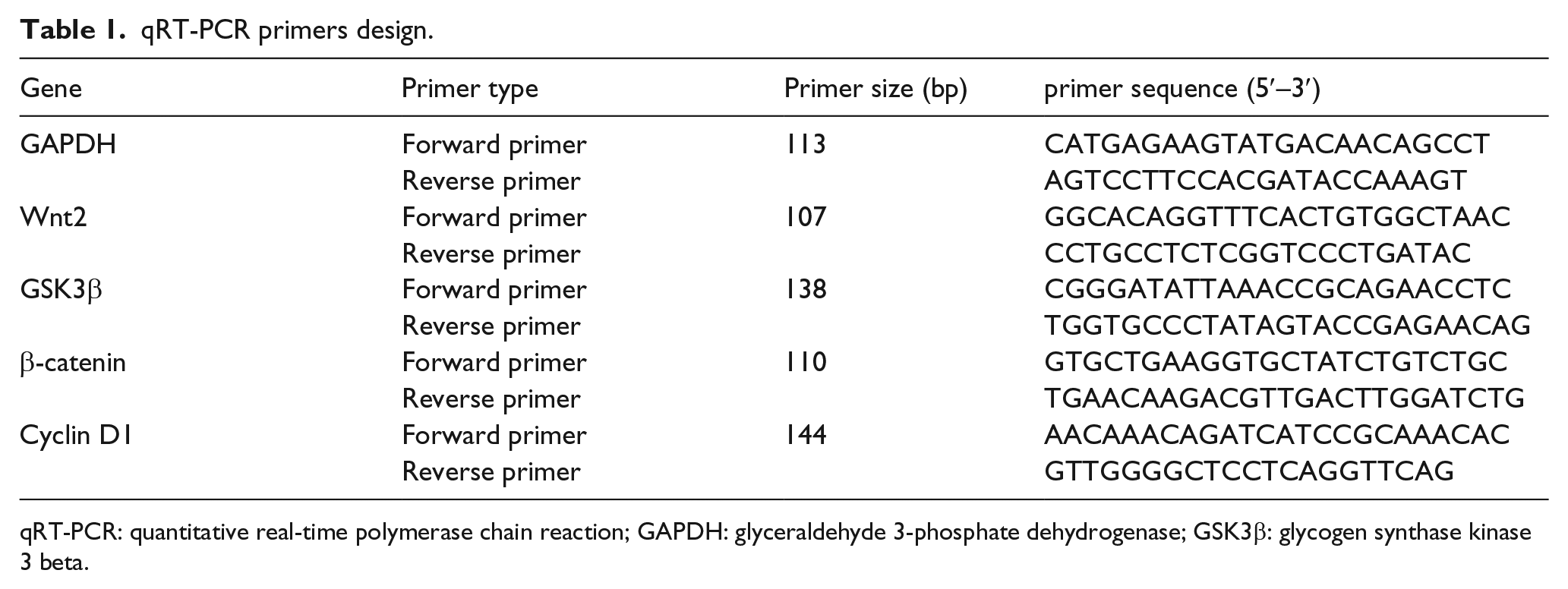
qRT-PCR: quantitative real-time polymerase chain reaction; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; GSK3β: glycogen synthase kinase 3 beta.
Protein extraction and Western blotting
Whole cells were lysed in a buffer (7 M urea, 1% Triton X-100, 100 mM dithiothreitol (DTT), and 20 mM Tris–HCl (pH 8.5)) and the protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). The proteins (10 µg) were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes (Protran; Whatman, Dassel, Germany). GSK3β (24198-1-AP) was obtained from Proteintech Group, Inc., (Chicago, IL, USA) and β-catenin (sc-7199), Wnt2 (sc-50361), p-GSK3β (sc-81494), cyclin D1 (sc-753), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; sc-365062) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); the secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA, USA). Western blotting was performed using standard methods as previously described. 12 The immunoreactive bands were visualized using the enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia, Buckinghamshire, UK), and relative band intensities were captured with the Imaging System (Syngene, Frederick, MD, USA).
Cell proliferation and cell cycle assay analysis
Cell Counting Kit-8 (CCK-8; Dojindo Molecular Laboratories, Kumamoto, Japan) assays were performed at 24, 48, 72, and 96 h after transfection to measure the effects of si-β-catenin and control on the viability of keloid fibroblasts. Cells were plated at 3000 cells per well in 96-well plates and 10 µL of the CCK-8 reagent was added to each well. The number of viable cells was estimated by Bio-Rad Microplate Reader (Bio-Rad Laboratories) at 450 nm. For cell cycle and apoptosis analysis, transfected cells in the log phase of growth were harvested, washed with PBS, fixed with 90% ethanol overnight at 4°C, and then incubated with RNase at 37° C for 30 min. A standard protocol for Annexin V–fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining was used and analyzed using a FACSCalibur Flow Cytometer (BD, San Jose, CA, USA) DNA histograms were constructed by the ModFit software and the results were presented as percentage of cells in a particular phase. Experiments were performed in triplicate.
Statistical analysis
The data were analyzed using SPSS 13 (SPSS, Chicago, Illinois, USA) and compared using one-way analysis of variance (ANOVA) with Fisher’s post hoc test. Data were presented as mean ± standard deviation (SD) of separate experiments (n ≥ 3). A p < 0.05 was considered to be statistically significant. All the experiments were performed at least in three independent assays under similar conditions.
Results
β-catenin was upregulated in human keloid fibroblast tissues
In order to confirm the dysregulation of β-catenin in keloid fibroblast tissues, we used qRT-PCR (the sequences of the primers are presented in Table 1) and western blotting to evaluate β-catenin expression, and we found that human keloid fibroblasts contained high levels of β-catenin messenger RNA (mRNA) and protein compared with normal age-matched skin cells (Figure 1(a) and (b)) and the expression data of other gene (Supplementary Figure 1). According to clinical diagnosis, all specimens were confirmed by pathological hematoxylin–eosin (H&E) staining in tissue sections (Figure 1(c)).

Upregulation of β-catenin in keloid tissue samples and their isolated normal age-adjusted skin samples. (a) A significant upregulation of β-catenin was demonstrated in human keloid tissue samples (keloid samples = 62, each sample total RNA agarose gel electrophoresis of the above were isolated from human keloid) when compared with non-keloid normal age-adjusted skin fibroblast tissues (normal samples = 21, each sample total RNA agarose gel electrophoresis of the below were isolated from human non-keloid) by qRT-PCR and the expression data of other gene (Supplementary Figure 1). (b) A significant upregulation of β-catenin was demonstrated in primary keloid fibroblast samples (n = 30) compared with non-keloid normal cells (n = 15) by western blotting (**p < 0.01 is significantly different from normal control tissues). (c) The degrees of fibrosis in normal skin (n = 15) and keloid tissues (n = 30) were detected by hematoxylin and eosin staining (H&E: 200×). Fibroblasts isolated from normal skin fibroblast tissues showed a remarkably lower staining intensity of keloid tissues.
Effective repression of β-catenin by delivering siRNA in fibroblast tissues
We next sought to inhibit β-catenin expression in primary fibroblast tissues using siRNA oligo, namely, β-catenin-homo-1, β-catenin-homo-2, and β-catenin-homo-3 targeting different sites of β-catenin transcript. The transduction efficiencies of all three targeting siRNAs as well as the scramble control were confirmed to be more than 90% in fibroblast cells by fluorescence microscopy (Figure 2(a)). Transfection of fibroblasts with β-catenin-siRNA significantly reduced the mRNA (Figure 2(b)) and protein levels of β-catenin (Figure 2(c) and (d)). In particular, β-catenin-homo-1 showed the strongest inhibition on both the β-catenin mRNA and protein expression. Therefore, the β-catenin-homo-1 was used in subsequent experiments.

Knockdown of β-catenin by delivering siRNA in keloid fibroblasts. (a) Transfection efficiencies of siRNA were confirmed to be over 90% by fluorescence microscopic detection of green fluorescence (FAM). (b–d) All three β-catenin-siRNA-oligos reduced β-catenin mRNA and protein expression as determined by (b) qRT-PCR and (c and d) western blotting. All the experiments were repeated three times (**p < 0.01 and ***p < 0.001 are significantly different from scramble negative control RNA oligo–transfected group).
β-catenin-siRNA inhibits fibroblast proliferation
Increased fibroblast proliferation directly contributes to excessive keloid fibroblast tissues. Effects of β-catenin-siRNA on fibroblast cell proliferation were measured by CCK-8 assay up to 4 days after transfection. Results revealed that β-catenin-siRNA reduced the number of viable fibroblasts in a time-dependent manner. The growth-inhibitory effect peaked on day 2–3 post-transduction (Figure 3(a)). To determine whether decreased number of viable cells was a result of cell cycle arrest, we analyzed the cell cycle distribution of β-catenin-siRNA or scramble control–transfected keloid fibroblasts by flow cytometry. At 48 h post-transfection, β-catenin-siRNA increased the proportion of keloid fibroblasts of G0/G1 phase when compared with the negative control. A reciprocal reduction of cells in S and G2/M phases was also observed in the β-catenin-siRNA group (Figure 3(b)). Loss of phosphatidylserine asymmetry is a molecular hallmark of apoptosis. In addition to determine whether β-catenin-siRNA induced apoptosis, phosphatidylserine externalization was assayed by flow cytometry of Annexin V–FITC/PI double-staining of the cells. As shown in Figure 3(c), the percentage of the Annexin V–positive apoptotic cells were significantly higher in fibroblasts transfected with β-catenin-siRNA oligo when compared with scramble control and blank controls. These data suggest that β-catenin-siRNA exerted its growth-arresting effect and affected apoptosis in keloid fibroblasts.

Knockdown of β-catenin suppressed keloid fibroblast proliferation and cell cycle progression. (a) The number of viable keloid fibroblasts was reduced upon β-catenin knockdown as determined by CCK-8 assay. (b) Flow cytometry was performed to analyze cell cycle distribution of keloid fibroblasts after propidium iodide staining. β-catenin knockdown induced G0/G1 phase cell cycle arrest. (c) Apoptosis was assayed by Annexin V–FITC/PI staining followed by flow cytometry. β-catenin knockdown did significantly alter the apoptosis. Data are from three independent experiments (*p < 0.05 and **p < 0.01 are significantly different from scramble negative control RNA oligo–transfected group).
Downregulating β-catenin to depress Wnt/β-catenin signal pathways
Wnt/β-catenin signaling is regulated at many levels, including some genes for cell proliferation, cell cycle and apoptosis, invasion and migration, and others. We therefore determined whether these two proteins could be key mediators in the process of fibroproliferative diseases, and thus measured the protein expression levels of β-catenin, Wnt2, p-GSK3β, GSK3β, cyclin D1, and those of which have been reported to be associated with cell phenotypes of keloid fibroblasts. As shown in Figure 4(a) and (b), β-catenin-siRNA reduced the protein expression of both Wnt2 and cyclin D1. Although β-catenin/Wnt2 were downregulated, the expression of GSK3 was not significantly decreased, the phosphorylation level was significantly decreased, p-GSK3 protein downregulation expression was observed.

Downregulation of fibrosis-related proteins in keloid fibroblasts by β-catenin knockdown. (a) Protein expression of β-catenin, Wnt2, GSK3β, phospho-GSK3β, and cyclin D1 in human keloid fibroblasts transfected with siRNA-β-catenin or control scramble RNA oligo was assessed by western blotting. (b) Protein expression in blank control cells was also examined. The expression of GAPDH was used for normalization to ensure uniform protein loading in each lane. All the experiments were repeated for three times (**p < 0.01 is significantly different from control scramble negative control RNA oligo–transfected group). (c) Proposed model of β-catenin regulation of keloid fibroblast phenotypes and the expression of fibrosis-related signaling mediators. Knockdown of β-catenin leads to decreased levels of Wnt2, phospho-GSK3β, and cyclin D1, thus resulting in lowered keloid fibroblast proliferation, cell cycle arrest in G0/G1 phase, and increased apoptosis of fibroblasts. The overall effect was that hypertrophic fibrosis of the keloid phenotype was suppressed.
Discussion
Though cell junctions are important to maintain tissue polarity and integrity, keloids are fibrotic tumors of the dermis that are form during a protracted and fibroproliferative diseases characterized by an exaggerated response to injury. This process significantly impairs the quality of life. Various studies have implicated that Langerhans cells, T cells, keratinocytes, and mast cells in skin fibrosis, as for other fibrotic disorders, but the pathological process has not been identified and there is no satisfactory treatment.3,13 Differences in gene expression in keloid fibroblasts may be due to either expression of the abnormal gene(s) or expression by another cell type to cause an epigenetic selection of fibroblasts in normal skin. Recent studies have provided evidence that epigenetic alterations occur during activation of wound healing and fibrosis, including increased expression of matrix metalloproteinase, plasminogen activator inhibitor, and cyclin-dependent kinase (CDK) inhibitors, and increased bone morphogenetic protein (BMP) signaling also has been implicated in fibrotic disorders. Manuel Berning et al. developed skin cancer models to dissect epidermal–dermal and tumor–stroma interactions. Fibroblasts isolated from adult skin self-assembled into dermal equivalents with their specific fibroblast-derived matrix that was stabilized by its own heterogeneous collagen fiber meshwork.14–16 The differential expression of several fibrosis-associated genes is confined to fibroblasts cultured from the keloid nodule. Soluble frizzled-related protein 1 (SFRP1) is a best known inhibitor of Wnt signaling, and increased Wnt signaling has been reported to be involved in the pathogenesis of keloids. Trichostatin A (TSA) blocks transforming growth factor beta–mediated myofibroblastic differentiation and induction of collagen gene expression in human skin fibroblasts.17–19
We focus on the effects of Wnt/β-catenin complexes in forming dysregulations in fibrotic diseases. β-catenin is a key regulator in the canonical Wnt signaling. The protein consists of a central region that is flanked by distinct N- and C-terminal domains. This scaffold serves as an interaction platform for many β-catenin-binding partners, at the membrane, in cytosol, and in the nucleus. The cytoplasmic β-catenin acts as an activator for T-cell factor, lymphoid enhancer factor, and transcription factors that result in a subset of cellular effects involving tissue morphogenesis, cellular adhesion, and tumor development. Nuclear β-catenin immunoreactivity suggests aberrant activation of subsequent epithelial–mesenchymal transition (EMT) in the pathogenesis of a number of fibrotic disorders.20,21 The β-catenin is also a critical element in the canonical Wnt signaling of tumorigenesis. Activating mutations of β-catenin or inactivating mutations of adenomatous polyposis coli (APC) or Axin have been found to be associated with a wide variety of human malignancies, such as colorectal, ovarian endometrial, heptocellular, desmoids, and pancreatic tumors. 22 Cancer studies suggest that deregulated β-catenin signaling promotes tumorigenesis by inducing expression of oncogenes such as c-Myc and cyclin D1.23,24 During melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC) development, the defective immune system together with altered mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription 3 (STAT3), and Wnt/β-catenin signaling pathways permits tumor growth. 25 Different cyclins exhibit distinct expression and degradation patterns which contribute to the temporal coordination of each mitotic event. Being a regulatory subunit of CDK4 or CDK6, its activity is required for cell cycle G1/S transition. 26 In response to cyclic adenosine monophosphate (cAMP), a series of events lead to phosphorylation of GSK3, though the mechanism seems different from the core Wnt/β-catenin pathway (phosphorylation leads to activation, not to degradation), the interaction between β-catenin and GSK3β is present and may have further evolved into the mode of action typical for canonical Wnt signaling. Without a Wnt signal, the levels of cytoplasmic free β-catenin are kept low. If not bound to E-cadherin, β-catenin is phosphorylated in the cytoplasm by the activity of a multiprotein destruction complex, marking β-catenin for degradation. Snail gene also is upregulated directly by β-catenin-mediated transcription, whereas Snail1 levels increase indirectly upon Wnt stimulation through blocking GSK3β activity and can directly interact with β-catenin and enhance Wnt target gene expression.27,28
Taken together, these data show that the β-catenin complex also plays a crucial role in modulating Wnt signaling and is involved in the pathogenesis of keloids. Our study demonstrated an orchestrated repression of profibrotic signaling and phenotypes (Figure 4(c)) upon delivery of β-catenin-targeting siRNA into keloid fibroblasts. Our findings not only further our understandings of the molecular mechanism underlying keloid formation but also enable clinicians to design and implement novel treatment strategies based on wound healing and keloid fibrotic disorders.
Footnotes
Acknowledgements
The authors thank Dr William for editing the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fujian Province Quanzhou Science and Technology Bureau of the Key Funding Projects (Grant No. 2012Z70) and Fujian Province Medical Innovation Funder (Grant No. 2009-CX-21).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.