
Abstract
Platelet-derived growth factor receptor has been implicated in many malignant and non-malignant diseases. Platelet-derived growth factor receptor-α is a tyrosine kinase and a side target for imatinib, a revolutionary drug for the treatment of chronic myeloid leukemia that has dramatically improved the survival of chronic myeloid leukemia patients. Given the importance of platelet-derived growth factor receptor in platelet development and its inhibition by imatinib, it was intriguing to analyze the role of platelet-derived growth factor receptor-α in relation to imatinib treatment in the development of imatinib-induced thrombocytopenia in chronic myeloid leukemia patients. We hypothesized that two known functional polymorphisms, +68GA insertion/deletion and −909C/A, in the promoter region of the platelet-derived growth factor receptor-α gene may affect the susceptibility of chronic myeloid leukemia patients receiving imatinib treatment to the development of thrombocytopenia. A case-control study was conducted among a cohort of chronic myeloid leukemia patients admitted to the Lok Nayak Hospital, New Delhi, India. A set of 100 patients of chronic myeloid leukemia in chronic phase and 100 age- and sex-matched healthy controls were studied. After initiation of imatinib treatment, the hematological response of chronic myeloid leukemia patients was monitored regularly for 2 years, in which the development of thrombocytopenia was the primary end point. Platelet-derived growth factor receptor-α promoter polymorphisms +68GA ins/del and −909C/A were studied by allele-specific polymerase chain reaction. Platelet-derived growth factor receptor-α messenger RNA expression was evaluated by quantitative real-time polymerase chain reaction. The messenger RNA expression results were expressed as 2−Δct ± standard deviation. The distribution of +68GA ins/del promoter polymorphism genotypes differed significantly between the thrombocytopenic and non-thrombocytopenic chronic myeloid leukemia patient groups (p < 0.0001). Moreover, +68GA del/del and ins/del genotypes in imatinib-treated chronic myeloid leukemia patients were associated with an increased risk of developing thrombocytopenia, with odds ratios 6.5 (95% confidence interval = 2.02–0.89, p = 0.001) and 6.0 (95% confidence interval = 2.26–15.91, p = 0.0002), respectively. Similarly, −909C/A promoter polymorphism genotype distribution also differed significantly between thrombocytopenic and non-thrombocytopenic chronic myeloid leukemia patient groups (p = 0.02), and a significantly increased risk of imatinib-induced thrombocytopenia was associated with −909C/A polymorphism mutant homozygous (AA) genotypes the odds ratio being 7.7 (95% confidence interval 1.50 to 39.91, p = 0.009). However, no significant risk of imatinib-induced thrombocytopenia was found to be associated with heterozygous genotype (−909C/A) with odds ratio 1.9 (95% confidence interval = 0.86–4.56, p = 1.14). Platelet-derived growth factor receptor-α messenger RNA expression was significantly higher in chronic myeloid leukemia patients compared to controls (p = 0.008). Moreover, patients with imatinib-induced thrombocytopenia had a significantly lower platelet-derived growth factor receptor-α messenger RNA expression, compared to patients without thrombocytopenia (p = 0.01). A differential expression of platelet-derived growth factor receptor-α messenger RNA was observed with respect to different +68 GA ins/del and −909C/A polymorphism genotypes. The +68GA deletion allele and −909A allele were significantly associated with lower expression of platelet-derived growth factor receptor-α messenger RNA. The platelet-derived growth factor receptor-α +68GA del/del, +68GA ins/del, and −909AA genotypes are associated with an increased risk of developing thrombocytopenia in imatinib-treated chronic myeloid leukemia patients. A significantly lower platelet-derived growth factor receptor-α messenger RNA expression accompanies the +68GA deletion allele in an allele dose-dependent manner. Platelet-derived growth factor receptor-α −909AA genotype is also associated with lower expression of platelet-derived growth factor receptor-α. The downregulation of platelet-derived growth factor receptor-α expression may play a causative role in imatinib-induced thrombocytopenia, a common side effect, in the subset of chronic myeloid leukemia patients with platelet-derived growth factor receptor-α +68 GA ins/del, +68 GA del/del, and −909C/A genotypes.
Keywords
Introduction
Chronic myelogenous leukemia (CML) is a clonal myeloproliferative disorder of hematopoietic stem cells (HSC). Initially, in the chronic phase (CP), myeloid progenitors and mature cells accumulate in the blood, bone marrow, and extramedullary tissues. 1 After 3–4 years, the disease progresses to advanced accelerated phase (AP) and terminates in blast crisis (BC) which is characterized by a maturation arrest in the myeloid or lymphoid lineage.2–4 Imatinib mesylate (IM), the first-line therapy for CML patients, induces a high rate of hematologic and cytogenetic responses in patients with CP CML. However, apart from delayed recovery of normal hematopoiesis, IM makes a significant number of these patients susceptible to many associated side effects. 5 The use of IM in CML has been linked with hematologic and non-hematologic toxicity. Hematologic side effects of IM are dose-dependent, reversible, and affect all hematopoietic cell lineages, though to a variable extent. Grade 3 or 4 neutropenia is among the most common dose-limiting effects, occurring in 14% of patients, followed by thrombocytopenia (8%) and anemia (3%). Thrombocytopenia is more common in the accelerated-phase CML and occurs following an average of 2 months of therapy. 6 These two hematologic adverse effects have been found to be associated with other tyrosine kinase inhibitors as well such as nilotinib and dasatinib though to a variable extent (Kantarjian et al., 2007). 7 Nonetheless, IM continues to be primary drug of choice for CML treatment and the only tyrosine kinase inhibitor being used in many countries including our country India. We tried to elucidate the molecular reasons underlying the variability of IM responses, which are still far from clear.
Platelet-derived growth factor receptor (PDGFR), given its role in the development and maturation of platelets, has been implicated in the pathogenesis of IM-induced thrombocytopenia. Platelet-derived growth factor (PDGF) is the principal mitogen in serum for mesenchymal cells and consists of a family of A, B, C, and D polypeptides which promote cell migration, proliferation, and survival by binding to their cognate homo- or heterodimeric tyrosine kinase receptors, PDGFRα and PDGFRβ. Prior to stimulation, PDGFRs reside primarily on the plasma membrane as inactive transmembrane proteins. In response to extracellular stimuli, PDGFRs dimerize and are autophosphorylated on tyrosine residues on the cytoplasmic face. This phosphorylation allows the PDGFR dimer to transmit the extracellular signal to cytoplasmic downstream molecules which carry this message to nucleus where different proteins acting as transcription factors are activated.8,9
The single-nucleotide polymorphisms (SNPs) and small insertion/deletions (ins/dels) within regulatory DNA sequences may directly affect the expression levels of nearby genes. Regulatory polymorphisms in multiple genes might not only decide the specific genetic characteristics of an organism but also the predisposition of that organism for complex diseases. Seven SNPs and one dinucleotide ins/del have been identified in the promoter region of the gene encoding the human platelet–derived growth factor receptor α (PDGFRα). The polymorphisms are linked, giving rise to five distinct haplotypes, namely, H1, H2α, H2β, H2γ, and H2δ. 10 Out of these, H1 and H2α are the two most common haplotypes and differ on six positions in PDGFRα promoter region from each other, while H2β, H2γ, and H2δ differ only on a single-nucleotide position from H2α.11,12 In vivo and in vitro assays have shown a higher promoter activity for H2α and H2β when compared to H1, H2γ, and H2δ haplotypes. This implies that the corresponding polymorphisms may also influence gene expression in vivo and thereby contribute to the susceptibility to PDGFRα-related diseases. 10
Deregulated PDGFRα expression has been found to be linked to neural tube defects (NTDs) in mice12,13 and humans.10,14 A two-base-pair insertion/deletion polymorphism (+68GA) and a SNP (−909C/A) in the promoter of PDGFRα have been previously identified, and in vitro assays have demonstrated decreased promoter activity with the respective deletion allele and A allele. Also, it has been shown by in vitro assays that −909C allele specifically binds to a protein complex which is present in all haplotypes except the low-activity haplotype H2γ. This protein complex is a member of the upstream stimulatory factor (USF) family of transcription factors. Moreover, a protein complex of 125 kDa specifically binds to the low-activity haplotype H1 at position +68GA del which may represent an H1-specific PDGFRα transcriptional repressor. 15 Therefore, in patients with the +68GA del and −909A alleles at PDGFRα gene locus, there may be decreased transcription of PDGFRα gene.
Given the possible role of the PDGFRα pathway in the developmental process of thrombocytes and the potential of polymorphisms in the promoter of PDGFRα to affect its transcriptional regulation, we examined whether the +68GA insertion/deletion and −909C/A polymorphisms in the promoter of PDGFRα contribute to the susceptibility to thrombocytopenia in CML patients treated with imatinib.
Methodology
Study design
Study design and inclusion/exclusion criteria of CML patients were planned carefully. Briefly, patients visiting medicine department of Lok Nayak Hospital, New Delhi, India, from August 2012 to June 2015 were screened for chronic myeloid leukemia on the basis of clinical criteria complete hemogram/bone marrow cellularity by the consulting physician and pathologist. The diagnosis of CML was further confirmed by molecular detection of BCR/ABL fusion transcripts by reverse transcription polymerase chain reaction (RT-PCR). Confirmed CML patients were approached and enrolled prospectively in the study after informed written consent was obtained from the subjects. Patients with other myeloproliferative disorders or patients with atypical CML were excluded from the study. Study design is illustrated in Figure 1. Patients were monitored every 15 days from the start of imatinib treatment, and details of the haemogram and other clinical parameters were noted. A case-control design was used with 100 CML patients in CP as cases and 100 age- and sex-matched normal healthy subjects as controls. Maulana Azad Medical College and associated Lok Nayak Hospital, New Delhi, Institutional Ethical Committee (IEC) approved the study.

Schematic diagram of patient selection and study design.
Data collection and definitions
Complete blood counts were monitored after every 15 days. Bone marrow biopsies and aspirates were performed at enrollment if indicated and every 3 months after assessing the clinical situation in detail. Samples were examined for cellularity, differential counts, and adequacy of megakaryocytes. Patients’ response was assessed following each hematological examination.
Definition of imatinib-induced thrombocytopenia
Imatinib-induced thrombocytopenia was diagnosed if the following three criteria were met:
Normal platelet count (150–450 × 109 L−1) before the beginning of imatinib treatment.
Fall in platelet count to <150 × 109 L−1 after 2 weeks or more of imatinib therapy not associated with other signs of CML disease progression, namely, increase in blast cells and basophils.
Fall in platelet count to <150 × 109 L−1 after 2 weeks or more of imatinib therapy not accompanied by fever or any other major systemic disorder.
Allele-specific polymerase chain reaction
+68 GA +/− and −909C/A polymorphisms were studied by allele-specific polymerase chain reaction (AS-PCR) technique. Briefly, genomic DNA was extracted from peripheral blood samples of CML patients and control subjects using DNA extraction kit (Gene Aid, India) following the manufacturer’s instructions. DNA concentration was determined by measuring the absorbance at 260 nm. The quality and integrity of the DNA were determined by the A260/280 ratio. DNA quality was also checked by ethidium bromide–stained 2% agarose gel electrophoresis pattern. The PDGFRα promoter polymorphisms were analyzed using allele-specific primers (+68GA ins/del: F1-5′GGGAGAGAAACAGA3′, F2-5′GGGAGAGAAACA3′, and R-5′GGAGGAGACTGC3′; −909C/A: F1-5′GGGTTCGACCCAC3′, F2-5′GGGTTCGACCCAA3′, and R-5′GCGGCGGGAGGG3′).
RNA isolation and reverse transcription
Total RNA, from blood samples of patients and control subjects, was extracted using TRIzol method. RNA concentration was determined by measuring absorbance at 260 nm. Complementary DNA (cDNA) synthesis kit (Thermo Fisher Scientific, USA) was used for cDNA synthesis according to the manufacturer’s instructions. Briefly, a mixture of total RNA (100 ng), 2 µL oligo (dT) primer, and 9 µL water was incubated in a thermal cycler for 5 min at 70°C and then for 10 min at 4°C. To this reaction mixture, 5× RT buffer (4 µL), 5 mM dNTPs (2 µL), RNAse inhibitor, and reverse transcriptase each 1 µL were then added in a total volume of 20 µL, and this final reaction mixture was incubated at 42°C for 1 h. After the reaction was complete, the mixture was heated to 95°C for 5 min to inactivate the reverse transcriptase and chilled at 4°C.
Quantitative real-time polymerase chain reaction
Quantitative RT-PCR (qRT-PCR) was carried out on a Rotor-Gene Instrument (QIAGEN; Skelton House, Lloyd, Manchester, UK) using 1 µL cDNA, equivalent to 100 ng RNA, for PDGFRα and β-actin genes’ expression with corresponding primers: PDGFRα-F (5′-AAAGAAGTTCCAGACCATCCC-3′) and PDGFRα-R (5′AGGTGACCACAATCGTTTCC-3′); β-actin-F (5′-CGACAACGGCTCCGGCATGTGC-3′) and β-actin-R (5′-CGTCACCGGAGTCCATCACGATGC-3′). A reaction mixture was prepared consisting of 10 µL SYBR Green Master Mix, 1 µL cDNA (100 ng RNA), 0.3 µL forward primer (25 pm/ µL), 0.3 µL reverse primer (25 pm/ µL), and 8.4 µL water in a 20 µL final reaction volume.
A standard QIAGEN Rotor-Gene protocol was established for simultaneous amplification of PDGFRα and β-actin genes. The first segment of the amplification cycle consisted of a denaturation program of 95°C for 10 min. The second segment consisted of three steps: denaturation (30 s at 95°C), primer annealing (30 s at 62°C), and elongation (45 s at 72°C) which was repeated for 40 cycles. The final segment consisted of a melting curve program (ranging from 35°C to 95°C).
Statistical analysis
Hardy–Weinberg equilibrium was tested using GraphPad Prism 5.lnk statistical software. A co-dominant model was chosen for computation of odds ratio (OR) for assessment of risk of developing thrombocytopenia in CML patients on imatinib treatment, taking CML patients without thrombocytopenia as the control group and CML patients with thrombocytopenia as the diseased group.
Statistical analysis was performed using chi-square or Fisher’s exact tests for categorical variables and one-way analysis of variance (ANOVA)/Mann–Whitney/Kruskal–Wallis test for continuous variables as appropriate using GraphPad Prism 5.lnk software package. A p value of <0.05 was taken as significant.
Results
Study population
From August 2012 to June 2015, suspected CML patients who were presented at Lok Nayak Hospital, New Delhi, were sent to the Leukemia Diagnosis Lab of Department of Biochemistry at Maulana Azad Medical College, New Delhi, for molecular detection of BCR/ABL fusion gene transcripts. Atypical CML patients were excluded from the study. The final cohort included a set of 100 chronic-phase CML patients and 100 age- and sex-matched healthy controls. Characteristics of the study population are shown in Table 1.
Demographic and clinicopathological features of CML patients.
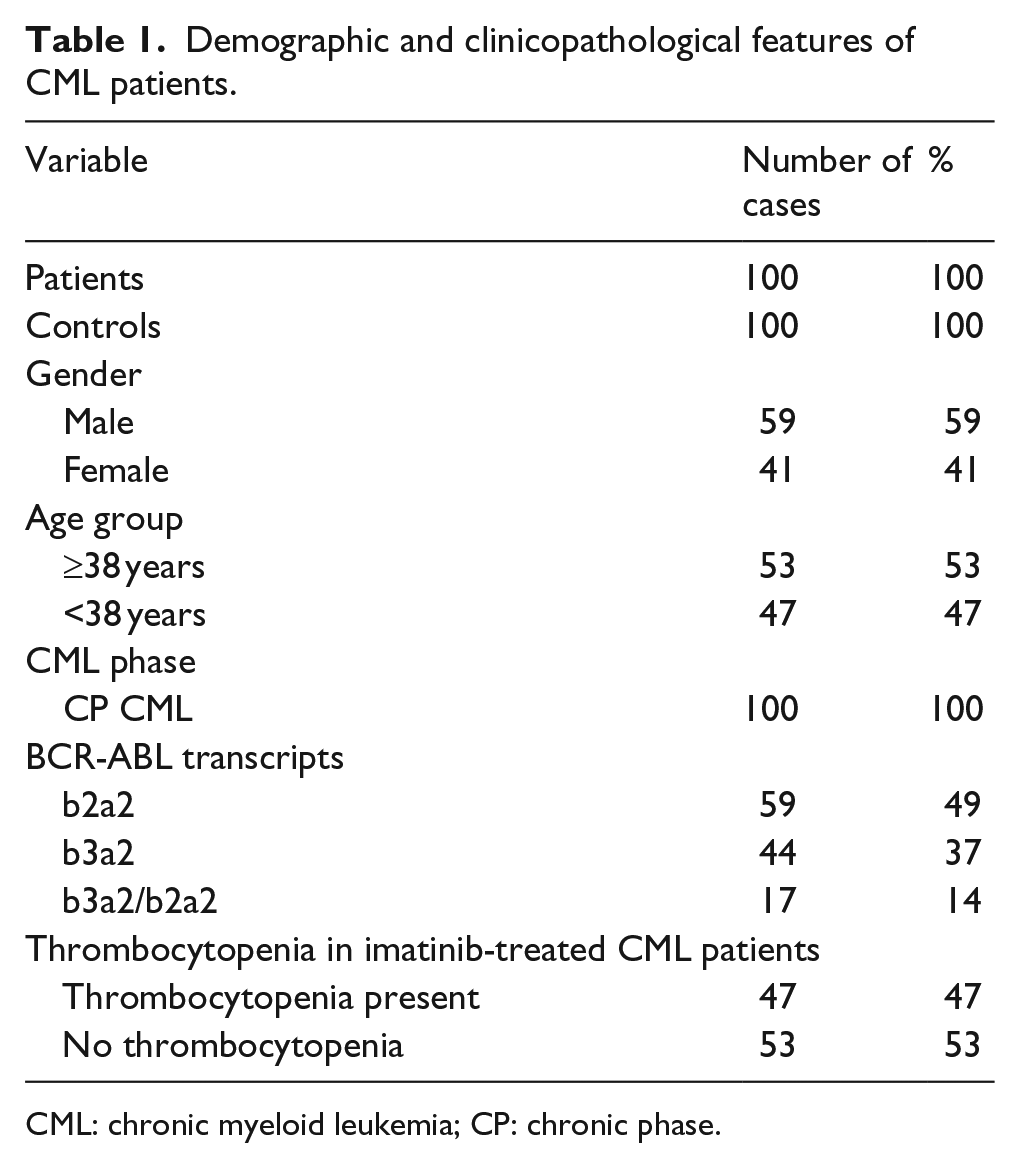
CML: chronic myeloid leukemia; CP: chronic phase.
Clinical course of thrombocytopenia in patients on IM
Of 100 CML patients, 47 developed thrombocytopenia within 3 months of initiation of IM (400–600 mg per day). Imatinib dose was then reduced in patients who developed thrombocytopenia on IM. The dose of IM (in mg/day) was averaged for each week of therapy by the treating physician for patients with thrombocytopenia and those without thrombocytopenia. Imatinib had to be stopped temporarily in some of the patients who developed severe thrombocytopenia. Doses of IM were reduced according to the results of bone marrow biopsy.
PDGFRα genotype association with imatinib-induced thrombocytopenia in CML patients: +68 GA del/del and ins/del genotypes in PDGFRα promoter are associated with a high risk of developing imatinib-induced thrombocytopenia in CML patients
In this case-control study, we examined the relation between the genotypes of PDGFRα promoter polymorphism at +68 GA position and imatinib-induced thrombocytopenia in CML patients. Compliance with Hardy–Weinberg equilibrium was confirmed among the control subjects. In all, 33 patients showed heterozygous (+68GA ins/del) genotype, 48 showed homozygous (+68GA ins/ins) genotype, and 19 showed homozygous (+68GA del/del) genotype, whereas out of 100 control subjects, 57 had heterozygous (+68GA ins/del) genotype, 18 homozygous (+68GA ins/ins) genotype, and 25 homozygous (+68GA del/del) genotype. This difference reached a good significance level statistically with a p value <0.0001 (Table 2 and Figure 2), which suggests that the +68GA ins/del PDGFRα polymorphism may influence the genetic susceptibility to the development of chronic myeloid leukemia.
Frequency of +68 GA ins/del polymorphism genotypes in CML patients and control subjects.

CML: chronic myeloid leukemia.

Electrophoretic gel image showing AS-PCR bands for PDGFRα +68GA ins/del polymorphism.
Importantly, the frequency of +68GA del/del and ins/del genotypes was more in patients who developed thrombocytopenia after imatinib treatment (28% and47%, respectively) as compared to those who did not (11% and 21%, respectively), and this difference was statistically significant with a p value <0.0001 (Table 3 and Figure 3).
Frequency of thrombocytopenia among imatinib-treated CML patients and +68GA insertion/deletion PDGFRα promoter polymorphism.

CML: chronic myeloid leukemia.
CML patient groups were highly statistically significant with a p value of 0.0001.

Frequency of +68 GA ins/del PDGFRα promoter polymorphism genotypes among CML patients who developed thrombocytopenia after imatinib treatment and those who did not. The differential genotypic distribution of +68 GA ins/del promoter polymorphism between the two groups was highly statistically significant with a p value of 0.0001.
Analysis of risk of thrombocytopenia associated with different +68 GA genotypes, using co-dominant inheritance model, indicated a significantly higher risk associated with +68 GA del/del homozygous genotype with OR of 6.5 (95% confidence interval (CI) = 2.02–0.89, p = 0.001) and +68 GA ins/del heterozygous genotype with OR of 6.0 (95% CI = 2.26–15.91, p ≤ 0.001). Among CML patients, +68GA del/del homozygous genotype was associated with slightly higher risk (OR = 6.5) for developing imatinib-induced thrombocytopenia as compared to +68GA ins/del heterozygous genotype (OR = 6). The OR was calculated by taking imatinib-induced thrombocytopenic CML patients as diseased group and CML patients who did not develop thrombocytopenia, after treatment with imatinib, as the control group (Table 4).
Risk of thrombocytopenia associated with +68ins/del PDGFRα gene promoter polymorphism genotypes.

CML: chronic myeloid leukemia; OR: odds ratio; CI: confidence interval.
Genetic association between −909C/A promoter polymorphism in PDGFRα gene and imatinib-induced thrombocytopenia in CML patients
Genotype frequencies of −909C/A PDGFRα gene promoter polymorphism did not differ significantly between CML cases and control group (p = 0.64; Table 5 and Figure 4). Significantly, the AA and CA genotypes were more frequent in imatinib-induced thrombocytopenic patient group (19% and 45%, respectively) compared to non-thrombocytopenic group (4% and 38%, respectively), and the difference reached a statistical significance with p = 0.02 (Table 6 and Figure 5).
Frequency of −909C/A polymorphism genotypes in CML patients and control subjects.

CML: chronic myeloid leukemia.

Frequencies of −909C/A PDGFRα promoter polymorphism genotypes among CML patients and age- and sex-matched healthy controls. No significant difference was observed between CML patients and healthy controls in the genotypic distribution of this polymorphism.
Frequency of thrombocytopenia in imatinib-treated CML patients and −909C/A PDGFRα promoter polymorphism.

CML: chronic myeloid leukemia; PDGFRα: platelet-derived growth factor receptor-α.
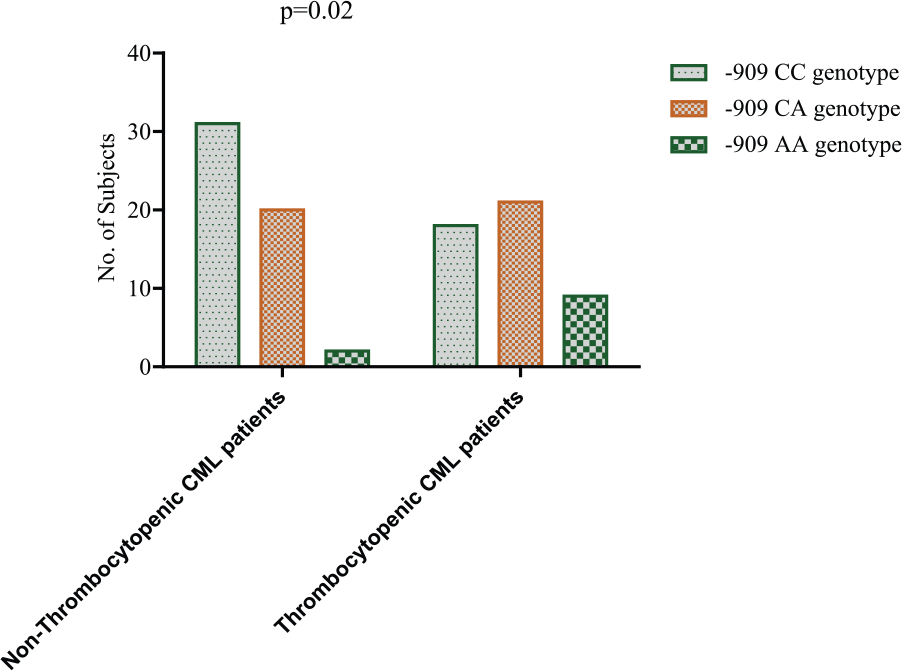
Frequency of −909C/A PDGFRα promoter polymorphism genotypes among CML patients that developed thrombocytopenia after imatinib treatment and those who did not. There was a differential distribution of −909C/A polymorphism genotypes among the two CML patient groups and this difference was statistically significant.
Analysis of risk of thrombocytopenia associated with different −909C/A genotypes, using co-dominant inheritance model, indicated a significantly higher risk associated with −909AA homozygous genotype with OR of 7.7 (95% CI = 1.50–39.91, p = 0.009). There was an increased risk associated with −909A/A genotype for the development of imatinib-induced thrombocytopenia but not with −909C/A genotype OR 1.9 (95% CI = 0.86–4.56, p = 0.14). The OR was calculated for the +68GA polymorphism (Table 7).
Risk of thrombocytopenia associated with −909C/A PDGFRα gene promoter polymorphism genotypes.

CML: chronic myeloid leukemia; OR: odds ratio; CI: confidence interval; PDGFRα: platelet-derived growth factor receptor-α.
Downregulation of PDGFRα gene expression during development of imatinib-induced thrombocytopenia
PDGFRα messenger RNA (mRNA) expression was evaluated by quantitative real-time PCR, and it was significantly higher in CML patients in comparison to healthy controls with a p value of 0.008 (Table 8 and Figure 6).
PDFRRα gene expression and thrombocytopenia in imatinib-treated CML patients.

CML: chronic myeloid leukemia; PDGFRα: platelet-derived growth factor receptor-α; SD: standard deviation.

qRT-PCR analysis of PDGFRα mRNA expression in CML patients and age- and sex-matched healthy control subjects. Expression level of PDGFRα was significantly higher in CML patients compared to normal healthy control subjects (p = 0.008).
We also observed a differential PDGFRα mRNA expression between two groups of CML patients: the one that developed thrombocytopenia after imatinib treatment and the other that did not. Thrombocytopenic CML patients had a significantly lower PDGFRα mRNA expression compared to CML patients who did not develop thrombocytopenia after imatinib treatment (p = 0.01; Table 8 and Figure 7).
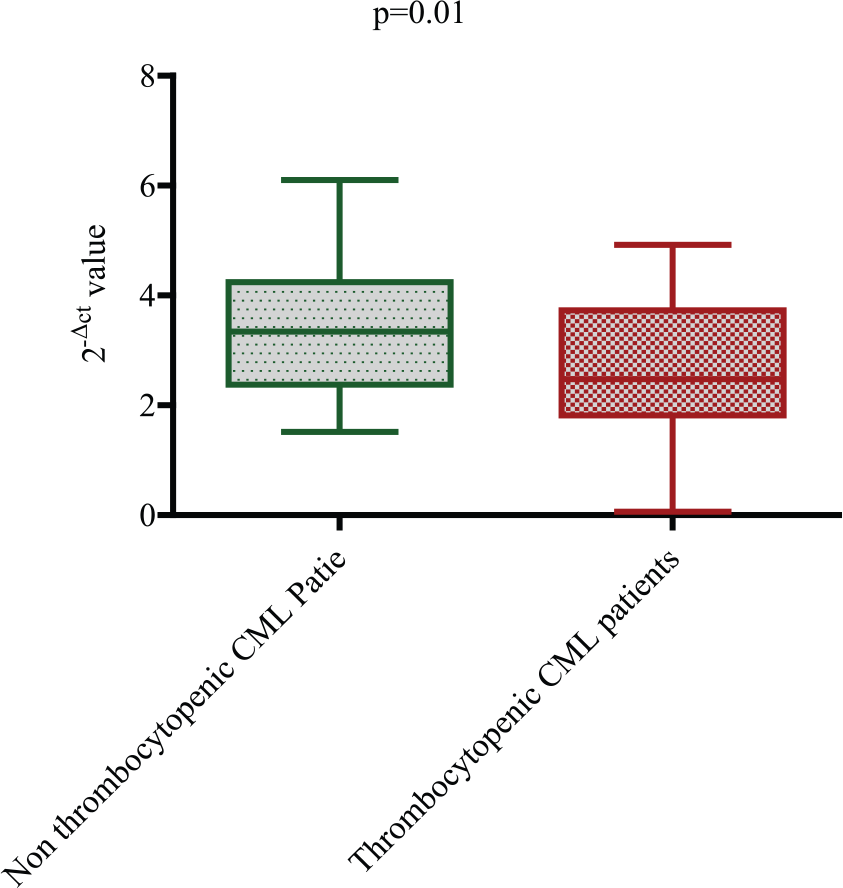
qRT-PCR analysis of PDGFRα mRNA expression in non-thrombocytopenic CML patients versus thrombocytopenic CML patients. A significant downregulation of PDGFRα mRNA expression was observed in thrombocytopenic CML patients compared to non-thrombocytopenic CML patients (p = 0.01).
Promoter polymorphisms in PDGFRα gene are significantly associated with its deregulated expression in CML patients
PDGFRα gene promoter region in humans contains 10 polymorphic sites that give rise to five distinct haplotypes, designated as H1, H2α, H2β, H2γ, and H2δ.10,14–16 These PDGFRα promoter haplotypes differ in their transcriptional activity and may thereby contribute to the pathogenesis of diseases with platelet dysfunction. We analyzed the PDGFRα expression with reference to genotypic distribution of two polymorphisms (−909C/A and +68 GA ins/del) in PDGFRα promoter in a group of 100 CML patients.
PDGFRα mRNA expression was compared in three groups of CML patients with reference to the −909C/A and +68 GA ins/del polymorphic sites, that is, homozygous for wild-type allele and heterozygous and homozygous for mutant allele as shown in Tables 9 and 10. A decreased mRNA expression of PDGFRα gene was associated with GA ins/del and GA del/del genotypes (2−Δct = 2.71 ± 1.07 and 2−Δct = 3.13 ± 1.35, respectively) as compared to GA ins/ins genotype (2−Δct = 3.39 ± 0.99), and this difference was statistically significant (p = 0.02; Table 9 and Figure 8). Similarly, the −909C/A polymorphism CC and CA genotypes were associated with higher PDGFRα mRNA expression (2−Δct = 3.55 ± 1.11 and 2−Δct = 3.27 ± 1.93, respectively) as compared to that of AA genotype (2−Δct = 2.57 ± 0.92). This difference also reached a good statistical significance with a p value of <0.0001 (Table 10 and Figure 9).
PDGFRα gene expression and +68GA insertion/deletion PDGFRα promoter polymorphism genotypes.

PDGFRα: platelet-derived growth factor receptor-α; SD: standard deviation.
PDGFRα gene expression and −909C/A PDGFRα promoter polymorphism genotypes.

PDGFRα: platelet-derived growth factor receptor-α; SD: standard deviation.

Differential expression of PDGFRα mRNA was observed in CML patients with different +68 GA ins/del genotypes. Maximum expression was associated with GA ins/ins genotype followed by GA ins/del and the lowest expression with GA del/del genotype (p = 0.02).

Differential expression of PDGFRα mRNA was observed in different −909 C/A genotypes. Maximum expression was associated with wild-type genotype CC followed by CA and the lowest expression with the AA genotype (p < 0.0001).
Discussion
Treatment of CML with imatinib is complicated by hematological toxicity in some patients, namely, anemia, neutropenia, and thrombocytopenia. The occurrence of these cytopenias is dependent on stage of CML and is more frequent in patients with accelerated phase/BC. These hematological adverse reactions usually occur within first few months of therapy and in many cases necessitate dose reduction or interruption of treatment for limited periods. Thrombocytopenia following imatinib therapy is a serious complication which may lead to uncontrolled internal/external hemorrhage and has to be carefully guarded against by frequent hematological examinations. A better understanding of molecular basis of thrombocytopenia in imatinib-treated CML patients is essential for evolving effective tools for management of this fairly common toxic side reaction.
SNPs in regulatory DNA sequences may influence gene expression by altering the binding affinity of transcription factors. Therefore, such SNPs may play a vital role in an individual’s susceptibility to different complex diseases. Platelet-derived growth factor receptor-α (PDGFRα) promoter in humans extends from −3600 to +118 position and contains a total of 10 polymorphic sites (8 SNPs and 2 insertion/deletion) that give rise to 5 distinct haplotypes, designated as H1, H2α, H2β, H2γ, and H2δ.10,15 H1 and H2α, which differ in 8 out of these 10 polymorphic positions,10,14–16 are the two most common haplotypes, with allele frequencies of 21% and 68%, respectively, in the Western European population.10,12,17 In this study, we have shown that the +68 GA PDGFRα promoter polymorphism depicts a statistically significant differential genotypic distribution among CML patients and healthy controls. The +68 GA deletion (−) allele, which is H1 haplotype specific, was under-represented in our cohort of CML patients. This may point toward an influence of PDGFRα +68GA ins/del on the susceptibility toward development of CML, but needs to be explored further in larger studies. In addition, there was an increased risk of developing imatinib-induced thrombocytopenia associated with +68 GA del/del and +68 GA ins/del genotypes. This is in parallel to the observation that H1 haplotype has more than 10-fold lower activity than H2α haplotype. The lower activity of the H1 allele has been suggested to be due to allele-specific DNA methylation and histone deacetylation, thus providing an epigenetic basis for the transcriptional repression. 15 In contrast, −909C/A PDGFRα promoter polymorphism did not show a statistically significant differential genotypic distribution among CML patients and healthy controls. However, the −909AA allele, which is H2γ haplotype specific, was associated with an increased risk of developing imatinib-induced thrombocytopenia although there was a significantly reduced PDGFRα expression associated with −909 AA genotype.
Polymorphisms in the promoter of the human PDGFRα gene have been found to be correlated with differential PDGFRα mRNA expression and risk of neural tube defects. 18 It has been shown that two of the polymorphic sites in the −3600 to +118 PDGFRα promoter region, that is, −909C/A and +68GA ins/del, show differential binding of nuclear proteins from the human osteosarcoma (HOS) cells, an in vitro model system for bone formation during axial development. The protein complex binding specifically to −909C allele but not −909A allele could be identified as a component of the upstream stimulatory factor 1 (USF1) family of transcription factors. USF proteins are ubiquitously expressed transcription factors of the basic-helix–loop–helix/leucine zipper (bHLH-zip) family and are encoded by two genes, USF1 and USF2. Both genes can give rise to two proteins by means of alternative splicing; USF1 and USF1/BD are encoded by the USF1 gene, whereas USF2a and USF2b are encoded by the USF2 gene.19–21 In contrast, the protein complex binding to +68GA del allele but not to +68GA ins allele has been characterized to have a molecular weight of approximately 125 kDa. The differential protein binding at these two sites is associated with the low promoter activities of both the H1 and H2γ haplotypes compared to H2α and H2β. A lower promoter activity for H2γ relative to H2α upon transfection into U2OS cells has also been observed. 14
Altered transcription of PDGFRα may result from a combination of promoter polymorphism and availability of transregulatory transcription factors. Hence, not only PDGFRα gene itself but also genes encoding upstream regulators of PDGFRα could be associated with the diseases related to aberrant expression of PDGFRα gene. In addition to USF1 that specifically binds to the PDGFRα promoter at the −909C position, it has recently been shown in PSFK-1 cells that the zinc finger protein ZNF148 can differentially bind the −1074C/A SNP, whereby it recognizes all haplotypes except H2δ. Interestingly, H2δ has been shown to be over-represented in patients with primitive neuroectodermal tumors (PNETs) and ependymoma brain tumors. 11 The low promoter activity of H1 compared to H2α is probably due to the +68GA del binding protein complex which potentially acts as an H1-specific repressor of PDGFRα transcription. Altered expression of PDGFRα has also been implicated in various neoplasias, including osteosarcomas, 22 chondrosarcomas, 23 gastrointestinal stromal tumors, 24 and glia brain tumors.25–29 We also observed an increased expression of PDGFRα gene in CML patients in comparison to healthy controls. It might be of great importance to study development of such tumors in the context of PDGFRα promoter haplotypes and upstream regulators of this gene.
To our knowledge, this study is the first study to show an association of PDGFRα gene expression with development of imatinib-induced thrombocytopenia in a subset of CML patients. Also, specific promoter polymorphism genotypes in PDGFRα gene (+68 GA ins/del, +68GA del/del, and −909C/A AA) were shown to be associated with an increased risk of the development of imatinib-induced thrombocytopenia in imatinib-treated CML patients.
Conclusion
The PDGFRα +68GA del/del, +68GA ins/del, and −909AA genotypes are associated with an increased risk of developing thrombocytopenia in imatinib-treated CML patients. A significantly lower PDGFRα mRNA expression accompanies the +68GA deletion allele in an allele dose-dependent manner. PDGFRα −909AA genotype is also associated with lower expression of PDGFRα. The downregulation of PDGFRα expression may play a causative role in imatinib-induced thrombocytopenia, a common side effect of imatinib treatment, in the subset of CML patients with PDGFRα +68 GA ins/del, +68GA del/del, and −909C/A AA genotypes.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study received funding from Indian Council of Medical Research (ICMR), New Delhi, India.