
Abstract
The ABL kinase inhibitor imatinib has been used as front-line therapy for Philadelphia-positive chronic myeloid leukemia. However, a significant proportion of imatinib-treated patients relapse due to occurrence of mutations in the ABL kinase domain. Although inhibitor sensitivity for a set of mutations was reported, the role of less frequent ABL kinase mutations in drug sensitivity/resistance is not known. Moreover, recent reports indicate distinct resistance profiles for second-generation ABL inhibitors. We thus employed a computational approach to predict drug sensitivity of 234 point mutations that were reported in chronic myeloid leukemia patients. Initial validation analysis of our approach using a panel of previously studied frequent mutations indicated that the computational data generated in this study correlated well with the published experimental/clinical data. In addition, we present drug sensitivity profiles for remaining point mutations by computational docking analysis using imatinib as well as next generation ABL inhibitors nilotinib, dasatinib, bosutinib, axitinib, and ponatinib. Our results indicate distinct drug sensitivity profiles for ABL mutants toward kinase inhibitors. In addition, drug sensitivity profiles of a set of compound mutations in ABL kinase were also presented in this study. Thus, our large scale computational study provides comprehensive sensitivity/resistance profiles of ABL mutations toward specific kinase inhibitors.
Introduction
The BCR-ABL fusion gene resulting from t(9;22)(q34;q11) chromosomal translocation was shown to be crucial for chronic myeloid leukemia (CML) pathogenesis. 1 This led to the approval of imatinib, a specific ABL kinase inhibitor, for the treatment of Philadelphia-positive (Ph+) CML. 1 However, emergence of secondary drug resistance due to point mutations in the imatinib-binding pocket of ABL kinase posed a significant clinical challenge.2,3 Several second- and third-generation inhibitors were developed and are used to treat imatinib-resistant CML patients. Notably, the drug sensitivity for individual point mutations varied against different ABL kinase inhibitors. 4 However, experimental and clinical drug sensitivity data were available for a limited number of most frequent ABL kinase mutations. 2 We thus computed ABL kinase mutant’s affinity toward various kinase inhibitors. In total, 1638 protein-inhibitor dockings were performed and drug sensitivity profiles for each of the 234 ABL mutants are presented. In addition, possible single/compound mutations that are resistant to the third-generation ABL kinase inhibitor ponatinib are predicted.
Materials and methods
RosettaBackrub software 5 was employed to generate mutant structures using dasatinib-bound ABL kinase (PDB ID: 2GQG) as the template. Wild type and mutant ABL kinase structures were imported into the Maestro. Modules of Schrodinger suite was used for protein and ligand preparation, molecular docking, and MM/generalized Born/surface area (GBSA) calculation. The protein preparation wizard was used to assign bond orders, add hydrogen atoms, and to optimize the hydrogen bond network at neutral pH. The protein was minimized with heavy atoms restrained to remain within a 0.3Å root mean square deviation (RMSD) of the original protein structure. Site map analysis was performed to determine the ligand-binding pocket (active site) and to calculate the size of binding cavity. A grid was generated around the binding site using GLIDE 5.6. The receptor van der Waals scaling for nonpolar atoms was set to 0.9 3 with a partial charge cutoff of 0.25. The kinase inhibitors were prepared for molecular docking using Ligprep module, and low energy conformers were selected for analysis. The inhibitors were docked into the generated grid using extra precision (XP) docking. The XP scoring function and docking protocol have been developed to reproduce experimental binding affinities. The XP Glide scoring function is presented in Equation 1. Equation 2 represents the terms that favor binding and equation 3 represents those that hinder binding. 6
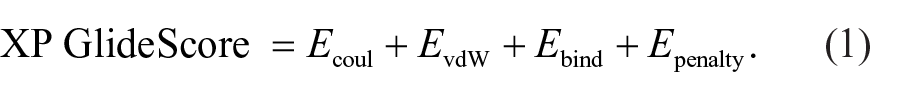
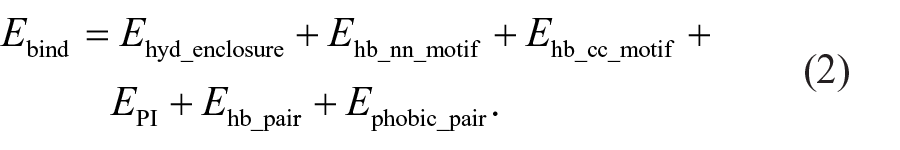

Protein-inhibitor complexes obtained from XP docking were subjected to Prime MM/GBSA calculation. Amino residues within 5Å of the ligand were allowed to flex, while minimizing the complex to ease minor steric clashes. Energies of the ligand, free protein, and the protein-lignad complex were calculated using OPLS-2005 force field and GBSA continuum solvent model. Binding free energy of the inhibitors in respective complexes is calculated based on the following equation:

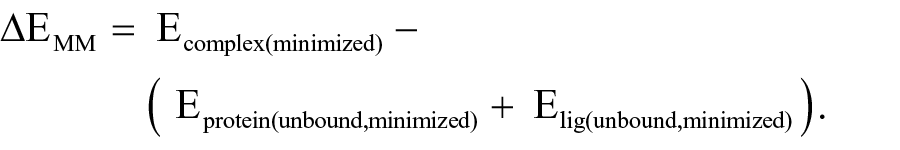
ΔGbinding is the calculated binding free energy, Ecomplex(minimized) is the MM-GBSA energy of the minimized complex, Eprotein(unbound,minimized) is the MM-GBSA energy of the minimized protein after separating it from its bound ligand, and Elig(unbound,minimized) is the MM-GBSA energy of the ligand after separating it from the complex and allowing it to relax. ΔEMM includes ΔEinternal (bond, angle, and dihedral energies), ΔEelectrostatic (electrostatic), and ΔEvdw (van der Waals) energies; ΔGGB is the electrostatic solvation energy, and ΔGSA is the nonelectrostatic solvation component. The polar contribution is calculated using generalized Born (GB), while the nonpolar energy is estimated using the solvent accessible surface area (SASA). The implicit solvent model estimates solvation free energies that implicitly include entropies associated with solvent.
Results and discussion
Point mutations that span entire ABL kinase were reported in newly diagnosed as well as imatinib-resistant CML patients (Figure 1). We performed a computational study to predict drug sensitivity profiles of 234 ABL kinase domain mutants (COSMIC (Catalogue of Somatic Mutations in Cancer) database) that were reported in CML patients (Figure 1). Binding affinity for each mutant–inhibitor interaction was calculated in XP mode of Schrodinger software using advanced scoring methods to eliminate false positives. A change in binding free energy that represents drug binding affinity for each docking event was recorded as an “xP score.” xP scores generated for most frequent imatinib-resistant ABL mutants (G250E, Q252H, Y253H, E255K/V and T315I) conformed to IC50 values from the published preclinical data (Supplementary Table 1). In addition to imatinib resistance, majority of these mutants displayed cross-resistance toward second- and third-generation ABL inhibitors (Table 1). Importantly, in line with earlier reports, G250E and E255V displayed high level of resistance to all the inhibitors tested (Table 1).7,8 Structural analysis of E255V mutant revealed the distortion of Y253 and K271 (Figure 2). Furthermore, G250E mutation caused a flip in the orientation of L248 and concomitant reduction of ligand-binding pocket size, thus resulting in incomplete binding of ABL inhibitors (Figure 3). Thus, loss of enzyme–inhibitor interactions and reduced ligand-binding pocket size are crucial factors contributing to inhibitor resistance.

Schematic representation of ABL kinase mutations analyzed in this study.
Validation of single point mutations with reference to published data.

xP scores of imatinib-resistant ABL mutants were compared to a validation data set for their conformity with existing preclinical and clinical data. Red—highly resistant (>25% decrease in xP score of the WT kinase); yellow—moderately resistant. ATP: adenosine triphosphate; WT: wild type.

Structural representation of effects caused by E255V mutation.

Schematic representation of effects caused by G250E mutation.
All inhibitors tested against wild type ABL kinase revealed higher binding affinity than that of the adenosine triphosphate (ATP) in the order: Dasatinib > Axitinib > Nilotinib > Ponatinib > Imatinib > Bosutinib > ATP (Table 1). As reported earlier, the T315I mutation showed significant cross-resistance toward all ABL inhibitors except for ponatinib 9 (Table 1). Analysis of top 30 predicted drug-resistant mutations for each ABL inhibitor exhibited variability in drug resistance profiles (Figure 4). Several of these 30 drug-resistant mutations were previously reported to cause resistance toward respective inhibitors: 16 for imatinib (G250V/F/E/A, Y253F, E255V/K/L, E258D, S265R, E282K, K291R, Q300R & T315I/D/P), 2 10 for nilotinib (G250A/E/R, E255K, V289F, F311V/L, T315N, M351K & E355A),10,11 14 for dasatinib (L248R, G250E/A, E255V/K/G/L, V299M, T315P/I/N & F317I/V/L),12,13 6 for bosutinib (L248V, G250E/A/R, E255V & T315I/A), 4 13 for axitinib (L248R, G250E/A, Q252K/H/R, E255V/G/K, F317V/I/S & H396P), 14 and 3 for ponatinib (G250E, Y253F and E255K/V) 15 (Figure 4).

Inhibitor resistance profiles of ABL kinase mutants.
Multiple mutations at G250, E255, and T315 residues constituted one-third of the top imatinib-resistant mutations (Figure 4); the gatekeeper mutation T315I showed the strongest imatinib resistance (Table 1 and Figure 4). Moreover, the topmost mutations found in this study (Figure 4) for dasatinib (F311V), 16 bosutinib (L248R), 4 axitinib (E255V), 14 and ponatinib (G250E) 15 were previously reported to display high level of resistance against respective inhibitors. Importantly, several of the top 20 imatinib-resistant mutations displayed sensitivity toward ponatinib (14), bosutinib (13), axitinib (8), dasatinib (7), and nilotinib (4), thus predicting the possibility of overcoming imatinib resistance (Table 2).
Computational analysis of top 20 imatinib-resistant mutations.

Inhibitor cross-resistance was studied by comparing drug sensitivity profiles for top 20 imatinib-resistant mutants toward second- and third-generation ABL kinase inhibitors: red—highly resistant (>25% decrease in xP score of the WT kinase); yellow—moderately resistant. WT: wild type.
In addition to single mutations, a set of previously reported compound mutations,17–19 when analyzed in the present study, 12 out of 17 mutations displayed imatinib resistance (Table 3). A recent study that published cellular IC50 values for a set of compound mutations 19 was taken as a reference to further validate the predicted drug resistance in this study. Nearly 70% of the predictions were in line with the earlier reported resistance profiles for ABL inhibitors (Supplementary Table 2). 19 Interestingly, 90% of the predicted dasatinib sensitivity for compound mutations correlated with the reference data set (Supplementary Table 2). Importantly, the compound mutation G250E-V299L displayed high level of cross-resistance against all the ABL inhibitors (Table 3 and Supplementary Table 2). In line with earlier reports, E255V-T315I showed ponatinib resistance, while T315I-M244V and T315I-Q252H remained ponatinib-sensitive, 19 thus indicating the usefulness of this approach also in predicting the drug sensitivity of compound mutations.
Drug sensitivity/resistance profiles of compound ABL kinase mutations.
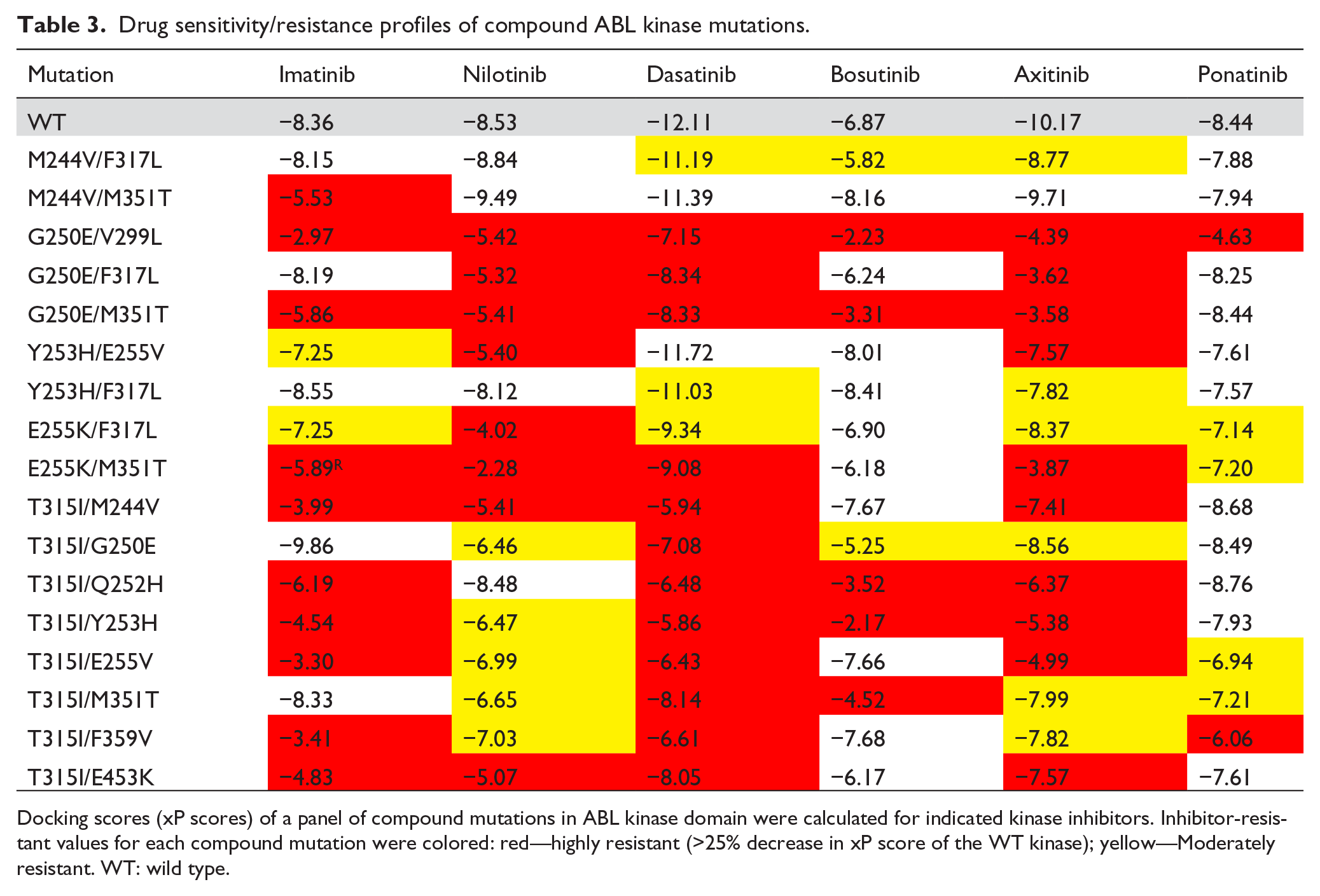
Docking scores (xP scores) of a panel of compound mutations in ABL kinase domain were calculated for indicated kinase inhibitors. Inhibitor-resistant values for each compound mutation were colored: red—highly resistant (>25% decrease in xP score of the WT kinase); yellow—Moderately resistant. WT: wild type.
Further analysis of inhibitor affinity profiles for multiple substitutions at hotspot residues revealed that G250E/V/F and T315I/P/D mutations are imatinib-resistant and G250A/R and T315A/N showed intermediate resistance (Supplementary Table 3). However, all mutations at T315 residue remained sensitive to ponatinib treatment (Supplementary Table 3). These findings are in line with previous observations, wherein variability in drug sensitivity among different amino acid substitutions at the gatekeeper (T315) residue was reported. Importantly, E255G/V showed resistance toward all ABL inhibitors (Supplementary Table 3). However, E255K/L mutants exhibited resistance to ABL inhibitors analyzed except for bosutinib (Supplementary Table 3). Further analysis revealed a reduction in size of the ligand-binding pocket in E255K/L mutants when compared with the wild type kinase (Supplementary Figure 1). As compared to imatinib, bosutinib, being a smaller ligand, is still able to bind deeply in to the reduced ligand-binding pocket of E255K/L mutants conferring inhibitor sensitivity (Supplementary Figure 1). Moreover, the number of interactions of imatinib with the mutant E255K/L is reduced as compared with that of the wild type kinase (Supplementary Figures 2–4). Specifically, imatinib binds wild type kinase via two hydrogen bonds (with Q252 and T315), two electrostatic interactions (with E286 and D325), and a π–π interaction with Y253 (Supplementary Figure 2). However, in mutants E255K/L, the number of interactions was reduced to one hydrogen bond (with Q252) and a π–π interaction (with Y253) (Supplementary Figures 3 and 4). On the contrary, the number of interactions of bosutinib with the mutant E255K/L is increased when compared to the wild type kinase (Supplementary Figures 2–4). Bosutinib showed only one hydrogen bond interaction (with T315) with the wild type kinase (Supplementary Figure 2). However, in E255K the number of interactions increased to two hydrogen bonds (with Q252 and D381), two electrostatic interactions (with Y253 and D381), and a π–π interaction (with Y253) (Supplementary Figure 3). In case of E255L mutation, bosutinib showed two hydrogen bond interactions (with Q252 and G249) and two charged electrostatic interactions (with E286 and D325). Overall, these results explain the heterogeneity in drug sensitivity/resistance among different substitutions at key amino acid residues.
A total of 1638 simulations were performed using seven ligands (ATP and six ABL kinase inhibitors) by docking over 234 ABL mutant structures (Supplementary Table 4). In addition to corroborating clinical findings, this study also predicts the potential of second- and third-generation ABL kinase inhibitors to overcome imatinib resistance caused by either single or compound mutations. However, drug resistance of a mutant defined in this study as a decrease in the binding affinity as compared to that of the wild type kinase, it is necessary to consider several other factors before extrapolating these findings to clinical setting. For example, a mutant with decreased binding affinity for a particular drug when compared to wild type kinase can still be inhibited at higher but clinically achievable concentrations. Despite the clinical utility of simulations presented here, it is still possible that a mutant predicted to be resistant might show sensitivity in the clinic and vice versa. Furthermore, the relative binding affinities of ATP and reversible ABL inhibitor toward each mutant need to be considered while classifying them as clinically sensitive or clinically resistant mutations. In conclusion, this study revealed variable degree of ABL inhibitor sensitivities (Supplementary Table 4) for a wide range of single/compound mutations that might be useful in selecting an effective treatment strategy.
Footnotes
Acknowledgements
S.K. and S.A. are co-first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is funded by the Start-up grant [No. F.4-5(136-FRP)/2014(BSR)] received (by RKK) from the UGC-FRP, Government of India.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.