
Abstract
Toll-like receptor 4 is overexpressed in various tumors, including cervical carcinoma. However, the role of Toll-like receptor 4 in cervical cancer remains controversial, and the underlying mechanisms are largely elusive. Therefore, Toll-like receptor 4 in cervical cancer and related mechanisms were investigated in this study. Quantitative reverse transcription polymerase chain reaction and western blot analyses were used to detect messenger RNA and protein levels in HeLa, Caski, and C33A cells with different treatments. Proliferation was quantified using Cell Counting Kit-8. Cell cycle distribution and apoptosis were assessed by flow cytometry. Higher levels of Toll-like receptor 4 expression were found in human papillomavirus–positive cells compared to human papillomavirus–negative cells. Proliferation of HeLa and Caski cells was promoted in lipopolysaccharide-stimulated groups but suppressed in short hairpin RNA–transfected groups. Apoptosis rates were lower in lipopolysaccharide-stimulated groups relative to short hairpin RNA–transfected groups. In addition, G2-phase distribution was enhanced when Toll-like receptor 4 was downregulated. Moreover, the pNF-κBp65 level was positively correlated with the Toll-like receptor 4 level in HeLa and Caski cells, though when an nuclear factor-κB inhibitor was applied to lipopolysaccharide-stimulated groups, the patterns of proliferation and apoptosis were opposite to those of the lipopolysaccharide-stimulated groups without inhibitor treatment. In conclusion, these data suggest that Toll-like receptor 4 promotes proliferation and apoptosis resistance in human papillomavirus–related cervical cancer cells at least in part through the Toll-like receptor 4/nuclear factor-κB pathway, which may be correlated with the occurrence and development of cervical carcinoma.
Keywords
Introduction
Human papillomavirus (HPV) is a sexually transmitted infection. Although infection rates vary between different regions, the rate tends to be highest among sexually active young women. More than 100 types of HPV have been identified to date. 1 Approximately 90% of HPV infections are eliminated by the body’s immune system, with only 1% developing into cervical cancer. 2 Nonetheless, cervical cancer presents a high mortality rate and is one of the most common gynecologic malignant tumors in the world. Persistent high-risk human papillomavirus (HR-HPV) infection, which mainly includes HPV 16 and HPV 18, is the major cause of the disease. The anatomic position of the cervix facilitates its long-term exposure to the microbial environment, which leads to hyperactivation of the inflammatory response. 3 However, the underlying mechanism needs to be further elucidated, and a more detailed exploration of the pathogenesis of cervical cancer is extremely urgent.
By recognizing conserved components of invading microbial pathogens, Toll-like receptors (TLRs) play a key role in the innate immune system. 4 TLR4, an important transmembrane pattern recognition receptor that recognizes exogenous ligands such as lipopolysaccharide (LPS), is a well-studied member of the TLR family in tumors and may participate in carcinogenesis and tumor progression during chronic inflammation by shaping the tumor microenvironment. 5 Indeed, a recent study reported that TLR4 is expressed in human cervical cancer HeLa cells at levels that are more than 100 times greater than other TLRs,6,7 an observation that provides proof of the link between TLR4 and the progression of cervical cancer. 5 However, the precise roles of TLR4 in tumor biology remain poorly understood. Evidence indicates the importance of both MyD88-dependent and MyD88-independent TLR4 signaling pathways, 8 which transmit signals via the phosphorylation of IκBα, an inhibitor of nuclear factor-κB (NF-κB). This phosphorylation event results in ubiquitination of IκBα followed by its degradation, leading to increased translocation of NF-κB to the nucleus where it eventually binds to the promoter region of target genes to activate transcription and promote tumourigenesis.9,10 Additionally, changes in NF-κB may contribute to the establishment of a molecular network in the pathogenesis of HPV infection. 11 Therefore, it has been suggested that TLR4/NF-κB signaling is involved in the occurrence and development of HPV-related cervical cancer.
In this study, we detected TLR4 expression in different cervical cancer cell lines and then upregulated and downregulated TLR4 levels in HPV-positive cell lines to examine changes in proliferation, apoptosis, and cell cycle distribution. In addition, we found that expression of pNF-κBp65 was positively correlated with the level of TLR4 in HPV-positive cells. When the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) was applied to LPS-stimulated cells, the patterns of proliferation and apoptosis were opposite to those of the LPS-stimulated groups without PDTC treatment. Taken together, our data show that TLR4 plays a crucial role in the carcinogenesis of HPV-related cervical cancer via the TLR4/NF-κB pathway, which is a potential target in the treatment of HPV-related cervical cancer.
Materials and methods
Cervical cancer cell lines and cell culture
Human cervical cancer cell lines HeLa (HPV18+), Caski (HPV16+), and C33A (HPV−) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 complete medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and maintained in 5% CO2 at 37°C. Cells were detached by routine trypsinization every 3–4 days. A Cell Counting Kit 8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). An Alexa Fluor 488 annexin V/Dead Cell Apoptosis Kit was obtained from Invitrogen (Carlsbad, CA, USA). PDTC, an inhibitor of NF-κB, was purchased from Abcam (Cambridge, MA, USA; catalog number: ab141406).
Short hairpin RNA transfection
Short hairpin RNAs (shRNAs) against human TLR4 were designed and synthesized by GeneChem Co., Ltd (Shanghai, China). Non-targeting shRNAs were used as negative controls (NCs; GeneChem Co., Ltd). At 24 h prior to transfection, cervical cancer cells were trypsinized (0.25% trypsin in DMEM) and seeded in six-well plates at a density of 4 × 105 cells/well. The cells were cultured in DMEM/F12 with 10% FBS for 24 h to reach 70% confluence, which was confirmed by inverted microscopy (CKX31 Inverted Microscope; Olympus Corporation, Tokyo, Japan). Cell transfection was performed using a Lipofectamine 3000 kit (Invitrogen) according to the manufacturer’s instructions. The experiments were performed in three groups, as follows: a TLR4-knockdown group transfected with different TLR4-shRNAs (TLR4-1, TLR4-2, and TLR4-3), an NC group (transfected with NC-shRNA), and an untransfected control group. After 24 h, the cells were processed using CCK-8 tests. After 48 h, the cells were collected for analysis of mRNA; after 72 h, the cells were harvested for western blotting and analysis of apoptosis and cell cycle distribution.
Quantitative real-time reverse transcription polymerase chain reaction
Total RNA was extracted using TRIzol (Invitrogen), and the reverse transcription reaction was performed according to the protocol from the PrimeScript RT reagent kit (Takara, Dalian, China). The polymerase chain reaction (PCR) amplifications were performed in a reaction with total volume of 10 µL, which included 5 µL FastStart Universal SYBR-Green Master (Rox), 3.6 µL double-distilled water, 1 µL complementary DNA (cDNA) template, and 0.2 µL each pair of forward and reverse primers. The TLR4 primer pairs for cDNA amplification were as follows: 5′-CCGAAAGGTGATTGTTG-3′ (forward) and 5′-AAGATGATACCAGCACGAC-3′ (reverse). The primers for amplification of β-actin were as follows: 5′-AAGGTGACAGCAGTCGGTT-3′ (forward) and 5′-TGTGTGGACTTGGGAGAGG-3′ (reverse). The PCR conditions included an initial denaturation step at 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 30 s. All experiments were carried out in triplicate and repeated three times. All samples were normalized to β-actin expression. The relative expression levels were calculated using the 2−ΔCT method.
Western blot analysis
Cells were harvested after corresponding treatments and washed with phosphate-buffered saline (PBS); cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) with a 1× protease inhibitor cocktail. The protein concentration was quantified using the bicinchoninic acid (BCA) protein assay kit (Beyotime) according to the manufacturer’s instructions. Equal amounts of protein (30 µg) were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane after electro-blotting under ice-cold conditions. The membrane was blocked for 1 h in Tris-buffered saline with Tween 20 (TBST) (25 mM Tris–HCl, pH 7.5, 137 mM NaCl, 2.7 mM KCl, and 0.05% Tween 20) with 5% non-fat milk at room temperature. After washing with TBST, the membrane was incubated overnight at 4°C with primary antibodies against TLR4 (1:500; Abcam, catalog number ab13556), NF-κBp65 (1:1000; Cell Signaling Technology, Boston, MA, USA, catalog number 6955S), pNF-κBp65 (1:1000; Cell Signaling Technology, catalog number 3033S), and β-actin (1:1000, Cell Signaling Technology, catalog number 3700S). After washing with TBST for 45 min (three washes of 15 min each), the membranes were incubated with the secondary antibody for 1 h at room temperature and then visualized using an enhanced chemiluminescence (ECL) system (ImageQuant LAS 4000, General Electric Company, Fairfield, CT, USA).
CCK-8
A CCK-8 kit was used to examine cell proliferation. Cells were seeded in 96-well culture plates at a density of 7 × 103 cells/well at 37°C. After 24 h, the medium was changed to a new pre-chilled medium, and the CCK-8 reaction solution (10 µL) was added to each well. The plates were incubated for 2–3 h. The absorbance was measured using a microplate reader (Multiskan MK3; ThermoFisher Scientific, Hudson, NH, USA) at a wavelength of 450 nm.
Assessment of apoptosis and the cell cycle
Treated cells were harvested, washed twice with PBS, and re-suspended in binding buffer at a concentration of 1 × 106 cells/mL, as described in the manufacturer’s instructions. Annexin V-fluorescein isothiocyanate (FITC) (5 µL) and propidium iodide (1 µL) were added to 100 µL of cell suspension, and the mixture was incubated for 20 min at room temperature in the dark. After adding 400 µL of binding buffer, the labeled cells were detected by flow cytometry within 1 h. Early apoptotic cells were detected using a FACS Calibur flow cytometer and subsequently analyzed using FlowJo 7.6 software. For cell cycle analyses, cells were fixed in 70% ethanol and stored at −20°C overnight; the cells were then labeled with propidium iodide (50 µg/mL) and RNase (100 µg/mL) for 30 min and analyzed by flow cytometry.
Statistical analysis
Values are expressed as the mean ± standard deviation (SD). All statistical analyses were conducted using Graphpad Prism 6.0 software (San Diego, CA, USA). Data were analyzed by t-tests. In all statistical comparisons, a p value of <0.05 was considered to be statistically significant.
Results
TLR4 is overexpressed in HPV-related cervical cancer cell lines
We examined the mRNA and protein expression of TLR4 in three cervical cancer cell lines using quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) and western blot analyses. As shown in Figure 1, compared to HPV-negative C33A cells, TLR4 expression was higher in HeLa and Caski cells, with slightly higher expression in Caski cells. The results demonstrated that HPV-positive cells exhibit higher expression of TLR4 than HPV-negative cells and that HPV16+ cells exhibit higher expression than HPV18+ cells. These findings suggest that TLR4 expression in cervical cancer cells may be associated with HPV infection, specifically HPV16.

TLR4 levels in three cervical cancer cell lines. (a) Protein expression of TLR4 was examined in HeLa, Caski, and C33A cells by western blotting. (b) Relative TLR4 mRNA levels were detected in HeLa, Caski, and C33A cells by RT-PCR.
TLR4 expression is increased in HeLa and Caski with LPS treatment
After administering 0, 1, 1.5, and 2 µg/mL LPS for 1 or 1.5 h, the level of TLR4 expression was detected by western blotting. In HeLa cells, TLR4 expression increased at low concentrations of LPS, especially at 1 µg/mL LPS for 1 h, but declined when LPS was increased to 2 µg/mL for 1 h (Figure 2(a)). In contrast, TLR4 expression increased in Caski cells treated with 2 µg/mL LPS for 1.5 h (Figure 2(c)). Overall, TLR4 expression increased in HPV-related cervical cancer cells with moderate LPS treatment. Similar to a previous study, C33A cells showed no response to LPS 6 (Figure 2(e)). We concluded that TLR4 expression is related to the HPV status of cervical cancer cells.
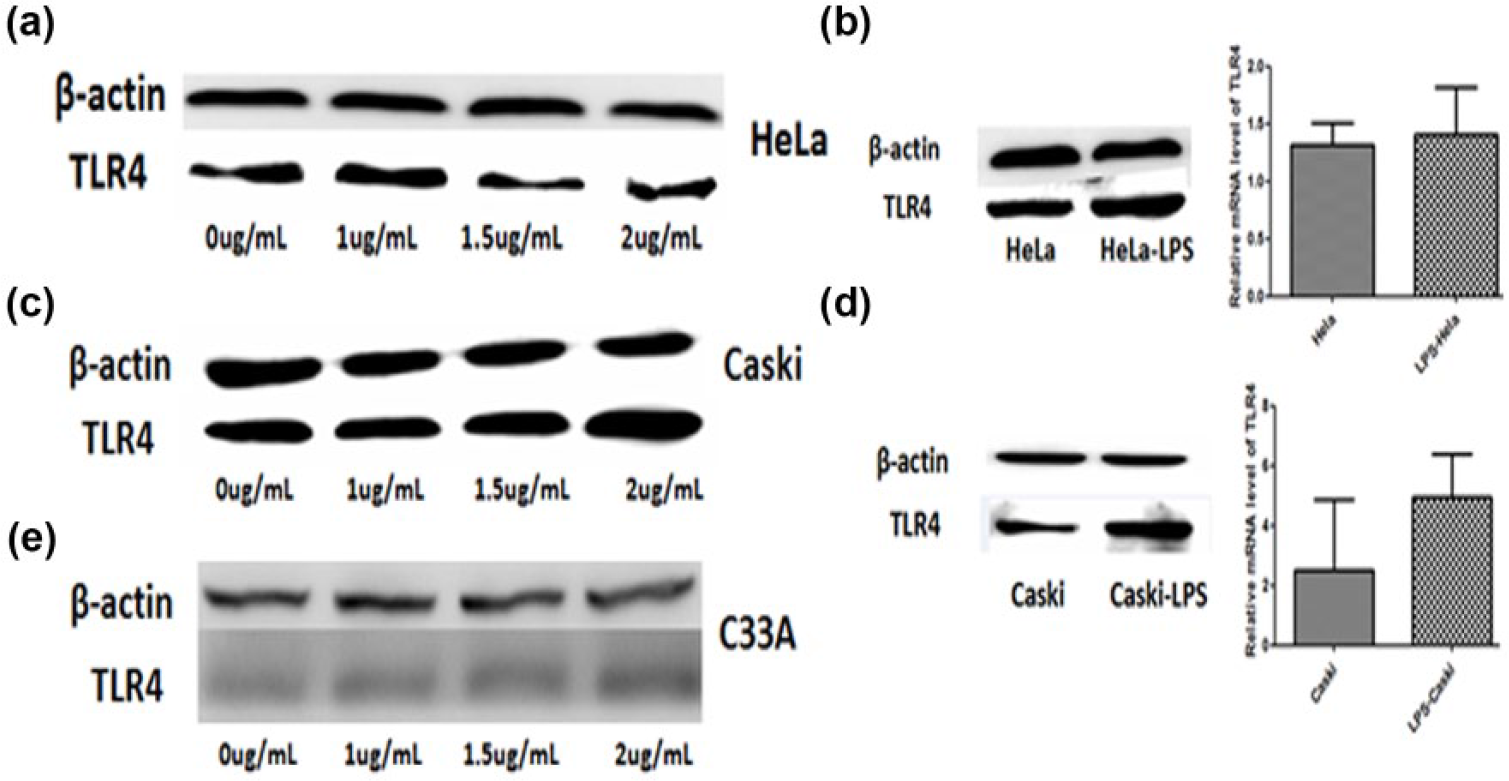
(a, c, e) Protein expression of TLR4 was examined in HeLa, C33A, and Caski cells exposed to different concentrations of LPS. (b and d) The level of TLR4 expression was detected in HeLa cells treated with 1 µg/mL LPS for 1 h and in Caski cells treated with 2 µg/mL LPS for 1.5 h.
TLR4 expression is downregulated in HeLa and Caski cells transfected with shRNA
To explore the biological significance of TLR4 in HPV-infected cervical cancer cells, we specifically knocked down TLR4 expression in HeLa and Caski cells using shRNA technology. The amount of TLR4 protein was significantly reduced in HeLa and Caski cells transfected with shRNA (TLR4-3), which showed efficient knockdown of TLR4 compared to shRNA (TLR4-1) and shRNA (TLR4-2) (Figure 3(a) and (c)). Additionally, there was a dramatic decline of TLR4 in both cell lines transfected with shRNA (TLR4-3) compared to the NC and control groups, but there was no significant difference between the NC and control groups (Figure 3(b) and (d)). Therefore, shRNA (TLR4-3) was used in the subsequent experiments.

Downregulation of TLR4 expression by shRNA in HeLa and Caski cells. (a and c) Protein expression of TLR4 was examined in control, TLR4-1, TLR4-2, and TLR4-3 shRNA groups by western blotting. (b, d–f) TLR4 expression in HeLa and Caski cells was examined in control, NC, and TLR4-3 shRNA groups by western blotting and RT-PCR.
LPS promotes proliferation and reverses TLR4-suppressed proliferation in both HeLa and Caski cells, whereas TLR4 depletion significantly inhibits cell cycle progression
LPS is an activator of TLR4, and this study demonstrated that TLR4 expression is upregulated in some cervical cancer cells. Treating HeLa and Caski cells with LPS (HeLa cells were treated with 1 µg/mL LPS for 1 h and Caski cells were treated with 2 µg/mL LPS for 1.5 h.) caused enhanced proliferation compared to the control groups (Figure 4(a) and (c)). We also examined the impact of TLR4 silencing on HeLa and Caski cell proliferation. Our data showed that downregulation of TLR4 by shRNA (TLR4-3) significantly reduced the proliferation of these cells compared to their NC counterparts (p < 0.05; Figure 4(b) and (d)). These results indicated that TLR4 promotes proliferation in HPV-positive cells. Moreover, cell cycle analysis revealed that TLR4 depletion caused significant inhibition of cell cycle progression, which led to selective accumulation of the cells in G2 phase compared to the NC groups (Figure 4(e) and (f)).

(a and c) The proliferation rate of HeLa and Caski cells treated with LPS. (b and d) The proliferation rate of HeLa and Caski cells transfected with TLR4-3 shRNA. (e and f) Significant G2-phase enrichment of HeLa and Caski cells with downregulated TLR4 expression (n = 3).
LPS treatment decreases the apoptosis rate, and TLR4 silencing increases the apoptosis rate in both HeLa and Caski cells
The effect of TLR4 on apoptosis was further explored in HeLa and Caski cells. The apoptosis rates of the HeLa control and LPS groups were 4.58 ± 0.135 and 1.22 ± 0.282, respectively (n = 3, p < 0.001) (Figure 5(a)), with similar results for Caski cells: 19.7 ± 2.69 and 6.92 ± 3.88, respectively (n = 3, p < 0.05) (Figure 5(c)). However, with TLR4 silencing, the results were opposite to those of the LPS group. For HeLa cells, the apoptosis rates of the NC and TLR4 silencing groups were 6.17 ± 0.374 and 7.30 ± 0.931, respectively (n = 3, p < 0.01) (Figure 5(b)), and the Caski cell apoptosis rate increased from 3.54 ± 0.37 to 15.57 ± 3.27 for the two groups (n = 3, p < 0.05) (Figure 5(d)). These results suggest that TLR4 promotes apoptosis resistance in HPV-positive cells.
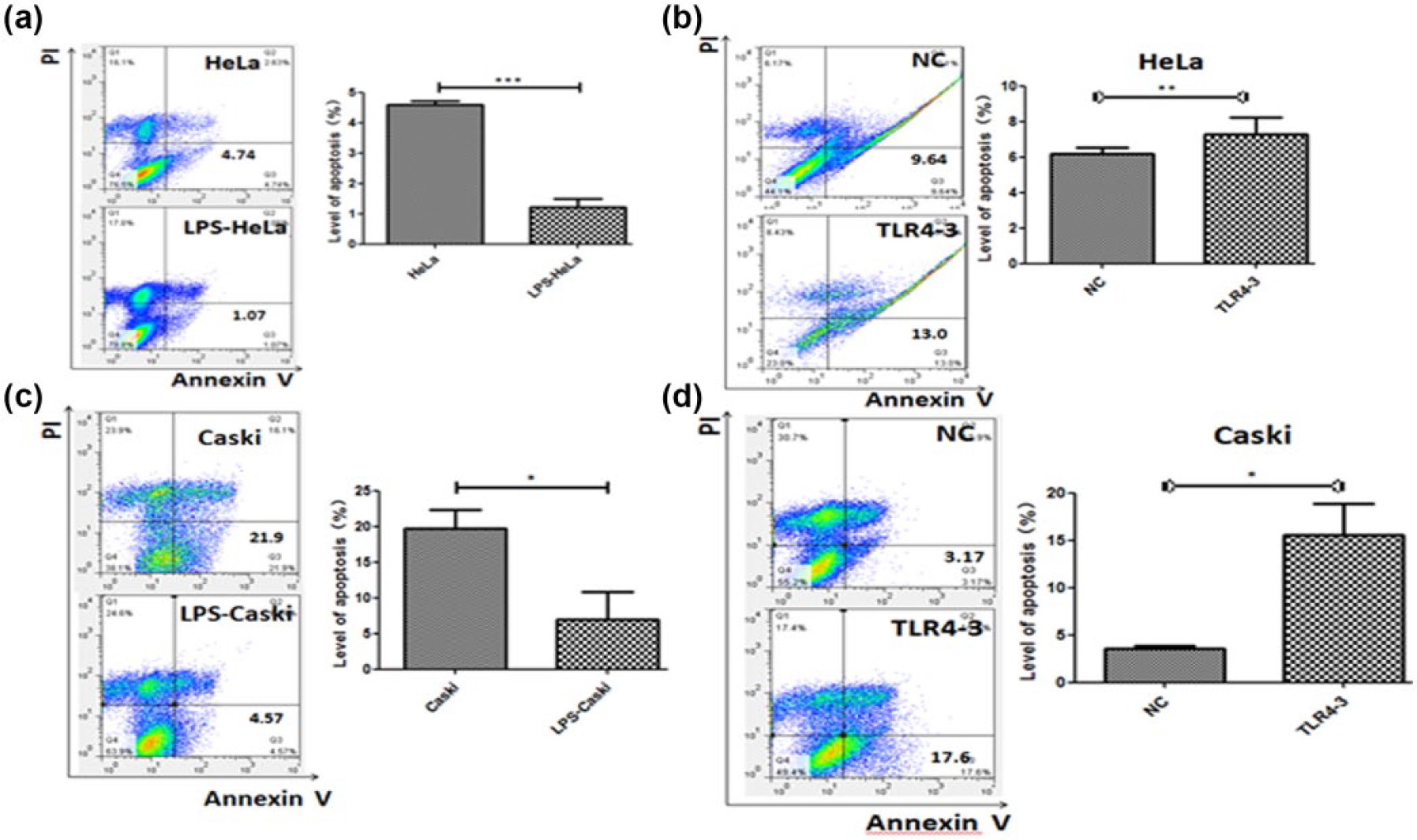
(a and c) Early apoptosis was decreased in HeLa cells treated with 1 µg/mL LPS for 1 h and Caski cells treated with 2 µg/mL LPS for 1.5 h. (b and d) Early apoptosis was increased in HeLa and Caski cells after TLR4-3 shRNA transfection for 72 h (n = 3).
Expression of pNF-κBp65 is positively correlated with TLR4 levels, and PDTC reverses these results in HeLa and Caski cells treated with LPS
Western blotting revealed no significant changes in NF-κBp65 expression in HeLa and Caski cells under different treatments. However, pNF-κBp65 was positively correlated with the TLR4 level (Figure 6(a)–(c)). Additionally, when PDTC, an inhibitor of NF-κB, was applied to the LPS groups, the results for proliferation and apoptosis were opposite to those of LPS-stimulated groups without any other treatment (Figure 6(d)–(f)). Indeed, the proliferation rates of HeLa and Caski cells treated with PDTC were reduced compared to groups without PDTC treatment (Figure 6(d)). The apoptosis rates for Caski cells treated with LPS, LPS + dimethyl sulfoxide (DMSO), and LPS + DMSO + PDTC were 0.526 ± 0.092, 1.44 ± 0.323, and 1.81 ± 0.301, respectively (n = 3, p < 0.001) (Figure 6(f)), whereas the rates for HeLa cells were 10.11 ± 1.62, 7.03 ± 1.07, and 5.40 ± 0.62, respectively (n = 3, p < 0.01) (Figure 6(e)), which was unexpected. These results indicated that NF-κB, a downstream signaling molecule of the TLR4 pathway, exerts its effects via phosphorylation in HPV-positive cells.

(a–c) Protein expression of NF-κBp65 and pNF-κBp65 was examined in HeLa and Caski cells treated with LPS or transfected with TLR4-3 shRNA for 72 h. (d–f) Proliferation and early apoptosis rate of HeLa and Caski cells treated with PDTC (n = 3).
Discussion
An increasing number of studies have found that TLR4 is expressed not only in immune cells but also in various types of cancer cells, including cervical cancer, gastric cancer, colorectal cancer, and non–small cell lung cancer (NSCLC) hepatocellular carcinoma (HCC) cells.12–16 Fukata et al. 17 showed that innate immune signaling by TLR4 shapes the inflammatory microenvironment in colitis-associated tumors. During the inflammatory response, TLRs, particularly TLR4, are involved in shaping the tumor microenvironment, which includes cancer, inflammatory, immune, stromal, endothelial, and epithelial cells. Cancer cells depend on their microenvironment to provide signals for growth, anti-apoptosis, angiogenesis, and metastasis, and mutual interactions between transformed cells and the microenvironment controls the tumor’s fate.6,18 Pimentel-Nunes et al. reported that TLR4 was overexpressed in normal gastric mucosa in patients infected with Helicobacter pylori. This study suggested that early exposure to an LPS environment reduced the expression of Toll-interacting protein (TOLLIP) and promoted the generation of inflammatory factors. The interaction between TLR4 and these inflammatory factors results in abnormal COX-2 transcription, which eventually promotes tumourigenesis. 19 In addition, it has been documented that expression of TLR4 is increased in hepatitis B virus (HBV)-related cirrhosis and HCC. 20 TLR4 expression is regulated in HCC cells at least in part by HBV infection, largely at the transcriptional level. 21 The anatomic position of the cervix allows for long-term exposure to the microbial environment, which leads to hyperactivation of the inflammatory response. Thus, HR-HPV infection promotes progression of the disease from low-grade lesions to invasive cervical cancer and plays an important role in tumourigenesis. 5 Regardless, the role of TLR4 in cervical cancer is still controversial. Yu et al. 22 showed that TLR4 was downregulated in high-risk, HPV16-positive cervical cancer cell lines, but Wang et al. 6 showed that TLR4 was overexpressed in HPV-positive cervical cancer cells, especially in HPV16-infected cells, but not in HPV-negative cells. Accordingly, in this study, we further confirmed the role of TLR4 in cervical cancer cells by upregulating and downregulating the level of this receptor. Our data revealed that TLR4 was expressed at higher levels in HPV-positive cervical cancer cells compared to HPV-negative cells. Additionally, the level of TLR4 was slightly higher in Caski (HPV16+) cells than in HeLa (HPV18+) cells, suggesting that TLR4 expression in cervical cancer cells may be associated with HPV infection and closely related to HPV16. In addition, TLR4 downregulation decreased proliferation and induced cell cycle arrest and apoptosis in HPV-infected cervical cancer cells. According to these data and the opposite results found in the LPS-stimulated group, we conclude that TLR4 is required for the survival and growth of HPV-infected cervical cancer cells. This study further confirmed the mediating role of TLR4 in the pathogenesis of HPV-related cervical cancer. Therefore, downregulating TLR4 may be an effective method to limiting the progression of HPV-related tumors in the future.
NF-κB, a transcriptional factor, is a downstream signaling molecule of the TLR4 pathway that controls the expression of numerous genes involved in inflammation.23,24 NF-κB is also a key molecule that mediates the relationship between inflammation and tumors. A previous study indicated that the TLR/MyD88/NF-κB signaling pathway plays a significant role in connecting inflammation and cancer invasion and progression. 25 In response to LPS, two pairs of intracellular adapter proteins, MyD88 and TIR-domain-containing adapter-inducing interferon-β (TRIF), interact with TLR4 to activate two main signaling pathways, MyD88- and TRIF-dependent. The MyD88-dependent pathway signal activates downstream mitogen-activated protein kinase (MAPK) pathways through interleukin (IL)-1 receptor–associated kinase (IRAK)-1, IRAK-4, and tumor necrosis factor (TNF) receptor–associated factor (TRAF)-6; these events in turn lead to activation of NF-κB. 26 In most unstimulated, normal cells, NF-κB is present in the cytoplasm as an inactive heterodimer composed of p50, p65, and IκB subunits. After activation, IκB undergoes phosphorylation- and ubiquitination-dependent degradation by the proteasome. As a result, the nuclear localization signals on the p50-p65 heterodimer are exposed, leading to nuclear translocation and binding to a specific consensus sequence to activate transcription of genes that encode growth factors and cellular invasion–related molecules. 27 Furthermore, some evidence indicates that NF-κB is constitutively activated in several types of tumors.28,29 In this study, we showed that pNF-κBp65 expression is positively correlated with the level of TLR4 in HPV-positive cervical cancer cell lines. When PDTC was applied to LPS groups, the results, including proliferation rates in both HPV-positive cell lines, tended to be opposite to those of LPS-stimulated groups without PDTC treatment. Apoptosis rates of Caski cells also tended to be opposite to those of LPS-stimulated groups without PDTC treatment, whereas the apoptosis rates of HeLa cells treated with PDTC and LPS also decreased, which was not consistent with the results we expected. These findings demonstrate that different HPV genotypes may employ different mechanisms of apoptosis resistance.
Overall, our experiments indicated that TLR4 promotes proliferation and apoptosis resistance in HPV-infected cervical cancer cells at least in part through the TLR4/NF-κB pathway, which may be correlated with the occurrence and development of cervical carcinoma.
However, the TLR4/NF-κB pathway is not the only pathway that affects the tumorigenesis of HPV-related cervical cancer, and our study showed that this pathway did not function in HeLa cell apoptosis. Thus, in the future, we will further investigate the possible pathways of TLR4’s involvement in HPV-related cervical cancer and their relationships. Moreover, a large body of evidence supports the close association of HR-HPV E6 and E7 with the pathogenesis of cervical cancer. A previous study indicated that the effect of HPV on TLRs may favor viral replication and thus survival in the host. 11 Previous research revealed a novel mechanism by which HPV16 suppresses the host’s immune response by deregulating TLR9 transcription. Interestingly, low-risk HPV E6 and E7 are unable to downregulate the TLR9 promoter. In contrast to TLR9 downregulation, the TLR3, TLR5, and TLR8 pathways are activated in HPV-infected human primary keratinocytes.11,30 Therefore, the relationship between HR-HPV and TLR4 in cervical cancer needs to be further elucidated.
In conclusion, our study showed that TLR4 promotes proliferation and apoptosis resistance in HPV-infected cervical cancer cells at least in part through the TLR4/NF-κB pathway, which is crucial to the tumorigenesis of HPV-related cervical cancer. Thus, the TLR4/NF-κB pathway may be a potential target for therapeutic agents in HPV-related cervical cancer. And further studies will be conducted to confirm the effect of PDTC in nude mice, which are injected subcutaneously with HeLa and Caski cells and visible tumors are developed, respectively.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China Grant 81272877.