
Abstract
High mobility group box 1 and toll-like receptor 4/myeloid differentiation factor 88 signaling pathway have been indicated to have oncogenic effects in many cancers. However, the role of high mobility group box 1/toll-like receptor 4/myeloid differentiation factor 88 signaling pathway in the development of gastric cancer remains unclear. In this study, we demonstrated that high mobility group box 1, toll-like receptor 4, and myeloid differentiation factor 88 were overexpressed in gastric cancer tumors compared with the adjacent non-tumor tissues. The overexpression of high mobility group box 1, toll-like receptor 4, and myeloid differentiation factor 88 were correlated with tumor-node-metastasis stage (p = 0.0068, p = 0.0063, p = 0.0173) and lymph node metastasis (p = 0.0272, p = 0.0382, and p = 0.0495). Furthermore, we observed that knockdown of high mobility group box 1 by high mobility group box 1-small interfering RNA suppressed the expression of toll-like receptor 4 and myeloid differentiation factor 88. Blockage of high mobility group box 1/toll-like receptor 4/myeloid differentiation factor 88 signaling by high mobility group box 1-small interfering RNA resulted in elevation of apoptotic ratio and inhibition of cell growth, migration, and invasion by upregulating Bax expression and downregulating Bcl-2, matrix metalloproteinase-2, nuclear factor kappa B/p65 expression, and the nuclear translocation of nuclear factor kappa B/p65 in gastric cancer cells. Our findings suggest that high mobility group box 1/toll-like receptor 4/myeloid differentiation factor 88 signaling pathway may contribute to the development and progression of gastric cancer via the nuclear factor kappa B pathway and it also represents a novel potential therapeutic target for gastric cancer.
Keywords
Introduction
Gastric cancer (GC) is a common lethal malignancy and one of the leading causes of cancer-related death worldwide, with approximately 951,600 people diagnosed with GC and 723,100 deaths in 2012. 1 Despite the advance in treatment for GC, the clinical outcome is unfavorable, with overall 5-year relative survival rates approximately 60% in Asian countries and 20% in Western countries. 2 Therefore, it is urgent to study the underlying molecular mechanism for gastric carcinogenesis, which is pivotal for identifying novel effective therapeutic targets.
High mobility group box 1 (HMGB1), a non-histone DNA-binding nuclear protein, is involved in both nuclear and extracellular activities. As a nuclear protein, HMGB1 functions as a chromatin-binding factor that bends DNA and facilitates access to transcriptional protein assembling on specific DNA targets such as nuclear factor kappa B (NF-κB) and p53.3,4 In addition to its nuclear role, HMGB1, which is actively secreted by various inflammatory cells and passively released from dying or necrotic cells, is implicated in inflammation, cell differentiation, cell migration, and cancer metastasis by acting as an extracellular damage-associated molecular pattern molecule (DAMP) binding to individual surface receptors, including the receptor for advanced glycation end products (RAGE) and toll-like receptors (TLRs) 2 and 4.5,6 HMGB1 overexpression has been demonstrated in many tumor types, such as hepatocellular carcinoma, colorectal cancer, bladder cancer, lung cancer, and GC.7–11
TLRs, a family of pattern recognition receptors that have important roles in innate immune defense, are involved in tumorigenesis.12,13 TLR4, which acts as a pivotal transmembrane pattern recognition receptor, recognizes various invading exogenous pathogens through pathogen-associated molecular patterns (PAMPs) and responds to DAMPs, 14 and the activation of TLR4 can stimulate the intracellular signaling cascade through a myeloid differentiation factor 88 (MyD88)-dependent or MyD88-independent pathway. 15 Recent reports indicated that TLR4 and its signaling cascade were believed to be implicated in tumor growth, progression, and invasiveness. 16 For example, TLR4 was involved in the promotion of hepatocellular carcinoma; 17 MyD88, the downstream adapter protein of TLR signaling, contributes to tumor progression in the ApcMin/+ mouse model; 18 TLR4/MyD88 signaling was associated with inflammation, tumor growth, and chemoresistance in ovarian cancer. 19
According to the previous studies, HMGB1, TLR4, and MyD88 are considered to play significant roles in carcinogenesis and tumor progression. However, little information is available on the role of HMGB1/TLR4/MyD88 signaling in GC. Therefore, in this study, we analyzed the expression of HMGB1, TLR4, and MyD88 in GC specimens and adjacent non-cancerous tissues, and investigated their clinical significance and these three protein’s correlation with each other. Moreover, we constructed HMGB1-small interfering RNA (siRNA), and transfected it into GC cells. The expression of HMGB1, TLR4, and MyD88, the bioactivity of GC cells and the effects of HMGB1 downregulation on downstream signaling pathways were observed to further explore the role of HMGB1/TLR4/MyD88 signaling in the pathogenesis of GC and the potential mechanism of this role.
Materials and methods
Patient samples
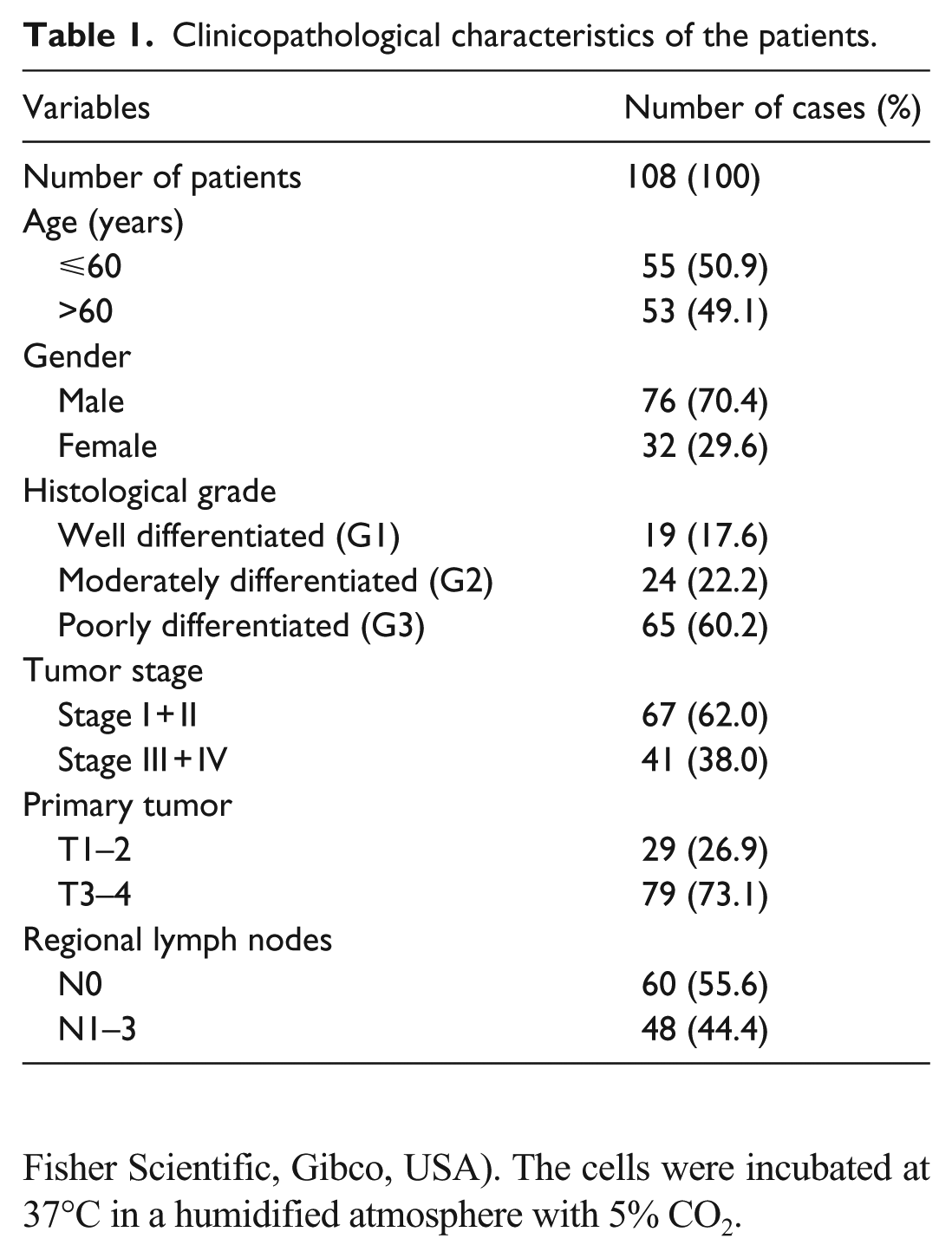
A total of 108 previously untreated primary gastric adenocarcinoma patients who received surgical resection in our department from January 2013 to October 2016 were enrolled in our study. All cases were confirmed with histological diagnosis. The pathologic tumor-node-metastasis (TNM) classification was made according to the criteria of the American Joint Committee on Cancer (AJCC). The research complied with patients’ consents and the approval from the Ethics Committee of Qilu Hospital, Shandong University. The detailed clinicopathological characteristics of these cases are summarized in Table 1.
Clinicopathological characteristics of the patients.
Immunohistochemical staining
A total of 108 patients’ paraffin-embedded cancer specimens and 30 adjacent non-cancerous tissues from these patients were obtained from the archive of Pathology Department of Qilu Hospital. The labeled streptavidin–biotin method was used to detect the expression of HMGB1, TLR4, and MyD88 in the specimens. After incubation for 1 h at 65°C, the sections were dewaxed in xylene and rehydrated in ethanol series. Then, the microwave antigen retrieval was performed using ethylenediaminetetraacetic acid (EDTA) antigen retrieval solution (pH 9.0; for HMGB1 and MyD88 detection) and citrate buffer (pH 6.4; for TLR4 detection). After that, the tissue sections were incubated with 3% H2O2 for 30 min to quench the endogenous peroxidase activity. After treated with goat serum at 37°C for 40 min, the slides were incubated overnight at 4°C with primary antibodies (HMGB1, 1:400; TLR4, 1:150; MyD88 1:300) (Abcam, USA), respectively. Then, sections were treated with biotinylated secondary antibodies for 30 min at 37°C and peroxidase-labeled streptavidin for 30 min at 37°C. Staining was visualized with DAB (3,3′-diaminobenzidine). Then, the slides were counterstained with hematoxylin, followed by dehydrated and mounted for microscopic observation.
Two experienced pathologists evaluated the immunostaining independently. The protein expression level was scored based on the staining intensity and percentage of positively stained tumor cells. The staining intensity was scored as 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (intense staining), and the percentage of positive staining as 0 (<5%), 1 (5%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (>75%). A weighted score was determined by multiplying the staining intensity score with percentage of positively stained tumor cells score, which ranged from 0 to 12. In addition, here, we defined the overexpression level of protein as the total score ⩾9. 20
Cell culture
The human GC cell lines MKN-28 and BGC-823 were obtained from the American Type Culture Collection (ATCC, USA). The cells were cultured in RPIM-1640 medium (GE Healthcare Life Sciences, Hyclone, USA). Both mediums were supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Gibco, USA). The cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Transfection of siRNA
The HMGB1-specific siRNA (HMGB1-siRNA) and a negative control (NC group) with no significant homology with human gene sequences were obtained from GenePharma (Shanghai, China). MKN-28 and BGC-823 cells in logarithmic growth phase were seeded into six-well plates, resulting in approximately 30%–50% confluency before transfection. After adherence for 24 h, 100 pmol of HMGB1-siRNA, or NC labeled with FAM, was added into 250 µL of Opti-MEM (Invitrogen), and 5 µL of Lipofectamine 2000 (Invitrogen) was added into 250 µL of Opti-MEM (Invitrogen, USA). After 5 min, the siRNA and Lipofectamine 2000 were mixed gently, and then added to the plates after 20 min. After incubated at 37°C for 4–6 h, cells were washed with phosphate-buffered saline (PBS) and then incubated with complete medium with 10% fetal bovine serum (FBS) until they were prepared for further assays. The sequences of siRNA and NC are as follows:
NC: Sense 5′-UUC UCC GAA CGU GUC ACG UTT-3
Antisense 5′-ACG UGA CAC GUU CGG AGA ATT-3′
HMGB1-siRNA: Sense 5′-CCU GUC CAU UGG UGA UGU UTT-3
Antisense 5′-AAC AUC ACC AAU GGA CAG GTT-3′
Western blot analysis
After transfected for 72 h, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with 1% protease inhibitors (Beyotime, China). Equal amounts of protein samples were separated on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). After that, proteins were transferred onto polyvinylidene fluoride (PVDF) membranes using a wet transfer apparatus (Bio-Rad, USA). Blots were blocked with 5% non-fat milk for 1 h at room temperature and then incubated overnight at 4°C with the primary antibodies HMGB1, TLR4, MyD88 (Abcam, USA), NF-κB/P65, Bcl-2, Bax, and matrix metalloproteinase-2 (MMP2; Cell Signaling Technology, USA), respectively. Subsequently, after washing for four times (5 min each) with Tris-buffered saline Tween 20 (TBST), the membranes were incubated with horseradish peroxidase-labeled secondary antibodies (Beijing Zhong Shan Biotech Co., Ltd., China) for 1 h at room temperature. Finally, the protein bands were visualized by enhanced chemiluminescence (ECL; Millipore, USA). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Proteintech, USA) protein was used as a control. Protein levels were quantified by Image J software.
5-Ethynyl-20-deoxyuridine assay and colony formation assay
Cell proliferation was analyzed by 5-ethynyl-20-deoxyuridine (EdU) assay kit (Ribobio, China). Cells were seeded into 24-well plates at a density of 5 × 104 cells per well and then treated with HMGB1-siRNA. After transfected for 48 h, the EdU assay was examined as described previously. 21 For colony formation, cells transfected with HMGB1-siRNA for 48 h were seeded into six-well plates at a density of 1 × 103 cells per well. After incubated for 12 days at 37°C with 5% CO2, colonies were fixed with precooled methanol and stained with 0.1% crystal violet for 20 min. The colony formation rate = (the colony number / the number of cell inoculation) × 100%.
Apoptosis assay
Flow cytometry was used to measure cell apoptosis using an Annexin V-FITC apoptosis detection kit (BestBio, China). Cells were harvested by trypsinization using trypsin without EDTA, washed two times with cold PBS, and resuspended in 400 µL of Annexin V binding buffer. Then, 5 µL of Annexin V-FITC and 10 µL of propidium iodide were added into the cell suspension, and incubated for 5 min and 15 min at 4°C in the dark, respectively. After that, cell apoptosis was detected with a FACScan flow cytometer (BD, USA).
Migration and invasion assay
Cell migration and invasion ability were assessed using the polycarbonate membrane (8-µm pore size) of a 24-well transwell chamber (Corning, Costar, USA). After transfected for 48 h, 5 × 104 cells suspended in 200 µL FBS-free medium were seeded into the upper chambers covered with 50 µL Matrigel (BD Biosciences, USA, 1:8 dilution; for the invasion assay) or without Matrigel (for the migration assay), and 600 µL complete medium with 10% FBS were added to the lower ones. After 24 h, the cells that had passed through the polycarbonate membrane were fixed with methanol and then stained with 0.1% crystal violet. At least five randomly selected fields were used to calculate the number of invaded cells under the microscope (Olympus, Japan).
Immunofluorescence assay
At 72 h after treatment, cells were washed with PBS, fixed in 4% formaldehyde, and permeabilized with 0.2% Triton X-100. After that, the fixed cells were treated in 1.5% normal goat serum and then incubated with a specific primary antibody for NF-κB/p65 (1:200 dilution) at 4°C overnight. After incubation with goat anti-rabbit IgG (H + L) labeled with Cy3 antibody (Beyotime; 1:500 dilution) for 1 h at room temperature in the dark, the nuclear DNA was stained with DAPI (4′,6-diamidino-2-phenylindole; Beyotime). A Zeiss Axioplan Universal microscope was used to analyze the cells.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 5.0 (GraphPad software, USA). A chi-square test was applied to explore the relationship between the overexpression of HMGB1, TLR4, and MyD88 and their clinicopathological characteristics. Spearman correlation was used to detect these three proteins’ relationship with each other. All experiments were independently performed at least three times. The results are presented as means ± standard deviation (SD). Statistical analysis was analyzed using Student’s two-tailed t-test. The p-values <0.05 were considered statistically significant.
Results
The expression of HMGB1, TLR4, and MyD88 in GC tissues
Immunostaining of HMGB1 protein was mainly observed in the nucleus. But in rare cases, the positive staining could be detected in nucleus and cytoplasm. Positive HMGB1 staining was detected in 29/30 (96.7%) adjacent non-tumor tissues (Figure 1(a)), and 107/108 (99.1%) cases of GC (Figure 1(b1) and (b2)) with no significant difference (p = 0.329, Table 2). But the proportion of HMGB1 overexpression (total expression score ⩾ 9) was increased in GC, compared with adjacent non-tumor tissues (44.4% vs 13.3%, p = 0.0019).

Detection of HMGB1, TLR4, and MyD88 immunoreactivity in adjacent non-tumor tissues and GC. (a), (c), and (e) The low expression of HMGB1, TLR4, and MyD88 were detected in adjacent non-tumor tissues, respectively. Strong immunostaining of HMGB1 was showed in cancer cells, localized in the (b1) nucleus or rarely in (b2) cytoplasm. Strong immunostaining of TLR4 was detected in cancer cells, localized in the (d1) membrane or (d2) cytoplasm. (f) Strong immunostaining of MyD88 was observed in cancer cells, which was localized in the cytoplasm. Original magnification 400×.
Expression of HMGB1, TLR4, and MyD88 in GC and adjacent non-cancerous tissues.

HMGB1: high mobility group box 1; TLR4: toll-like receptor 4; MyD88: myeloid differentiation factor 88; GC: gastric cancer.
p < 0.01.
Immunostaining of TLR4 protein was observed in the membrane and cytoplasm. A total of 29/30 (96.7%) adjacent non-tumor tissues (Figure 1(c)) and 108/108 (100%) cases of GC (Figure 1(d1) and (d2)) showed TLR4 expression with no significant difference (p = 0.0569). However, the rate of TLR4 overexpression was elevated in GC, compared with adjacent non-tumor tissues (35.2% vs 10%, p = 0.0076).
Immunostaining of MyD88 protein was mainly observed in the cytoplasm. MyD88 expression was detected in 28/30 (93.3%) adjacent non-tumor tissues (Figure 1(e)), and 108/108 (100%) cases of GC (Figure 1(f)) with significant difference (p = 0.0069). The proportion of MyD88 overexpression was also increased in GC, compared with adjacent non-tumor tissues (41.7% vs 13.3%, p = 0.0041).
Clinicopathological significance of HMGB1, TLR4, and MyD88 expression
The clinicopathological data for HMGB1, TLR4, and MyD88 are shown in Table 3. The overexpression of HMGB1, TLR4, and MyD88 were correlated with TNM stage (p = 0.0068, p = 0.0063, p = 0.0173) and lymph node metastasis (p = 0.0272, p = 0.0382, p = 0.0495). A high co-expression of HMGB1/TLR4/MyD88 was also found to be significantly associated with TNM stage and lymph node metastasis (p = 0.0077, p = 0.0017).
Clinicopathological features according to HMGB1, TLR4, and MyD88 expression.

HMGB1: high mobility group box 1; TLR4: toll-like receptor 4; MyD88: myeloid differentiation factor 88; GC: gastric cancer.
p < 0.05; **p < 0.01.
The correlation among HMGB1, TLR4, and MyD88 expression in GC
HMGB1 overexpression was observed in 29/38 (76.3%) and 29/45 (64.4%) of high TLR4 and MyD88 expression tissue, respectively, while the rate in low TLR4 and MyD88 expression tissue was 19/70 (27.1%) and 19/63 (30.2%), respectively. TLR4 overexpression was detected in 25/45 (55.6%) of high MyD88 expression tissue, while the rate in low MyD88 expression tissue was 13/63 (20.6%). Based on the Spearman correlation analysis, of these three proteins, there was a positive correlation with each other: HMGB1 and TLR4 (r = 0.4336, p < 0.0001), HMGB1 and MyD88 (r = 0.3402, p = 0.0003), and TLR4 and MyD88 (r = 0.3605, p = 0.0001; Table 4).
Correlation among HMGB1, TLR4, and MyD88.
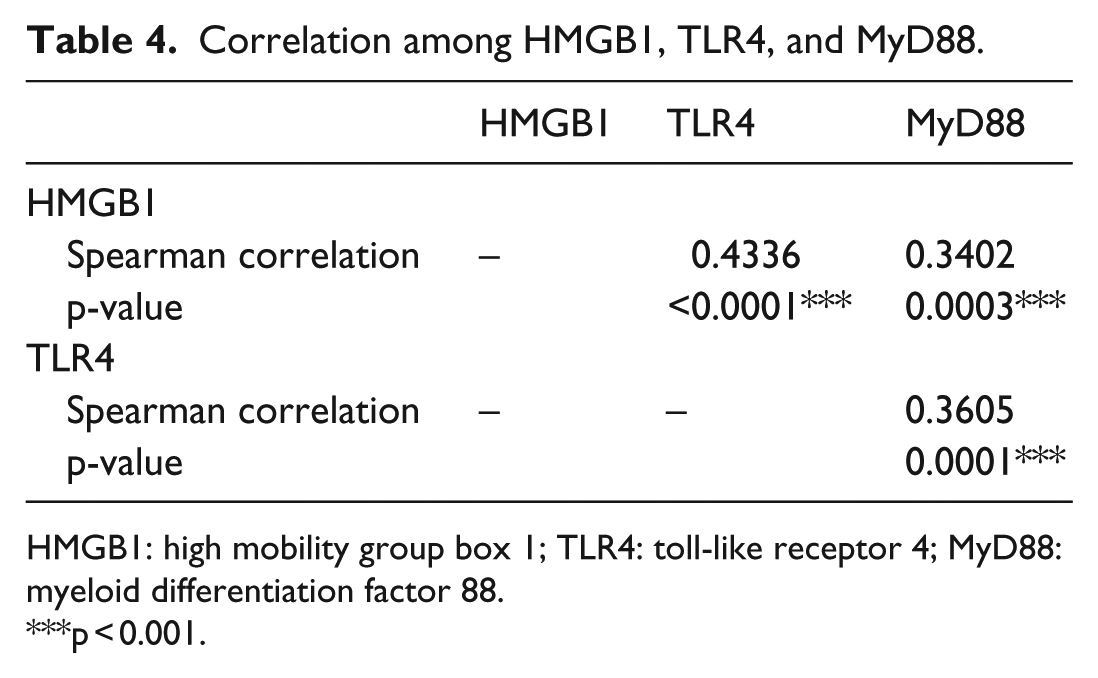
HMGB1: high mobility group box 1; TLR4: toll-like receptor 4; MyD88: myeloid differentiation factor 88.
p < 0.001.
RNA interference effectively inhibits HMGB1 expression in GC cells
For the following experiments, two GC cell lines (BGC-823and MKN-28) were divided into two groups: the NC group (siNC-transfected cells) and the siRNA group (HMGB1-siRNA–transfected cells). Western blot was used to detect HMGB1 expression after cells transfected with HMGB1-siRNA. The results showed that the expression of HMGB1 protein in the siRNA groups was significantly inhibited than that in NC groups (p < 0.01; Figure 2(a)), indicating that the RNA interference (RNAi) was effective.
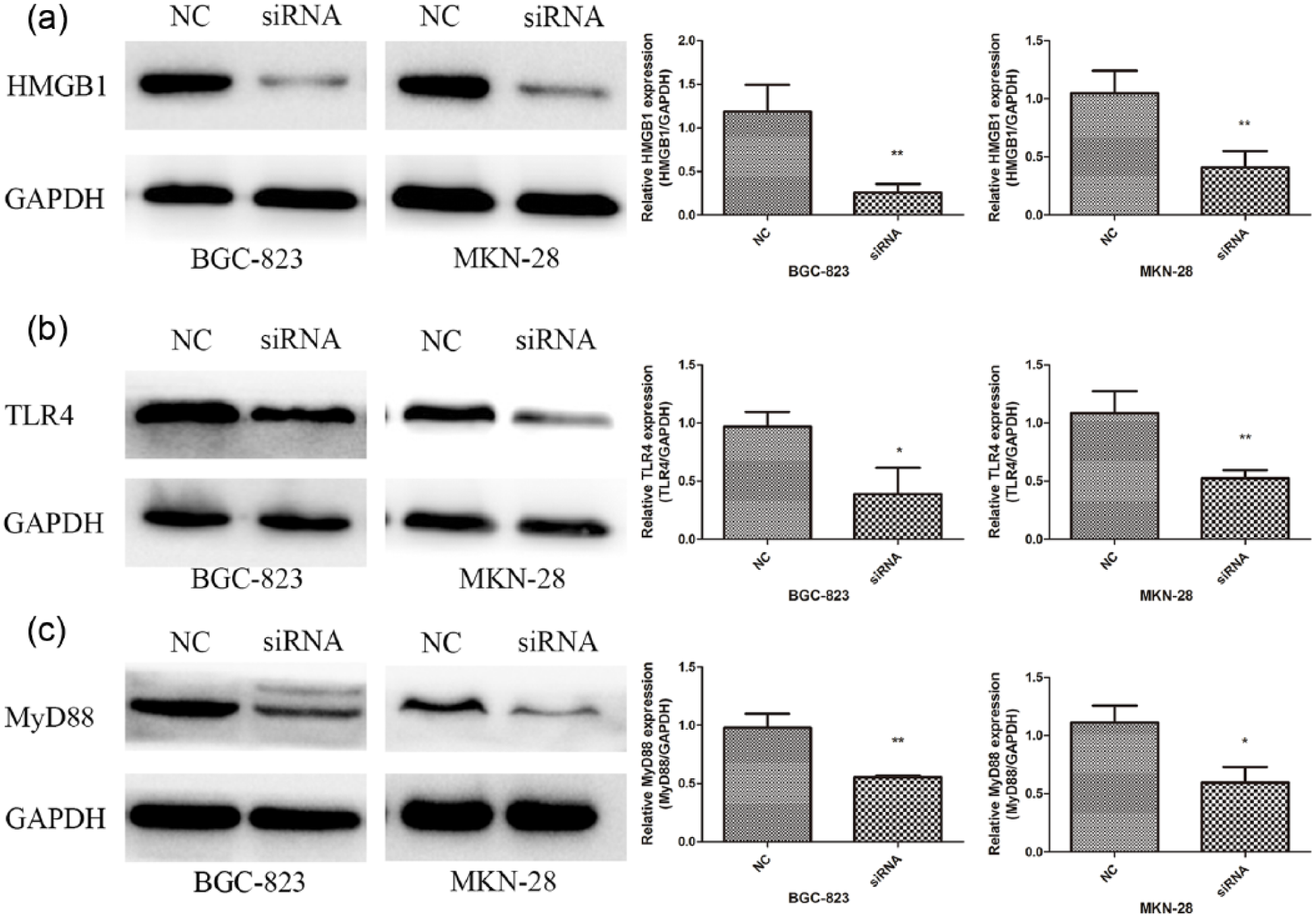
Effects of HMGB1-siRNA on the expression of HMGB1, TLR4, and MyD88 in BGC-823 and MKN-28 cells. Western blot was used to detect HMGB1, TLR4, and MyD88 expression. (a) Relative HMGB1 protein expression. (b) Relative TLR4 protein expression. (c) Relative MyD88 protein expression. Significantly decreased expression of HMGB1, TLR4, and MyD88 was found in HMGB1-siRNA–transfected cells. The data are presented as the means ± SD of three independent experiments.
HMGB1 knockdown mediates TLR4 and MyD88 downregulation
Extracellular HMGB1 involves in inflammation, cell differentiation, cell migration, and cancer metastasis by binding to individual surface receptors, such as TLR4, leading to the activation of NF-κB and mitogen-activated protein kinases (MAPKs) intracellular signaling.5,6 Based on previous studies, we assumed that TLR4 signaling might mediate the function of HMGB1 on the development of GC. To identify this hypothesis, western blot was performed to measure the expression of TLR4 and MyD88 using the HMGB1-siRNA. As shown in Figure 2(b), the expression of TLR4 was suppressed in siRNA groups compared with NC groups (BGC-823 p < 0.05; MKN-28 p < 0.01), and in Figure 2(c), the expression of MyD88 was also downregulated in HMGB1-siRNA–transfected cells compared with the siNC-transfected cells (BGC-823 p < 0.01; MKN-28 p < 0.05).
HMGB1 knockdown inhibits GC cell proliferation
Cell proliferation ability was analyzed using an EdU incorporation assay. The results displayed that the number of EdU positive cells in the HMGB1-siRNA groups was significantly reduced compared with the NC groups (p < 0.001; Figure 3(a)). For the colony formation assay, the data showed that HMGB1-siRNA–transfected cells formed significantly smaller and fewer colonies compared with the siNC-transfected cells (BGC-823 p < 0.001; MKN-28 p < 0.01; Figure 3(b)).
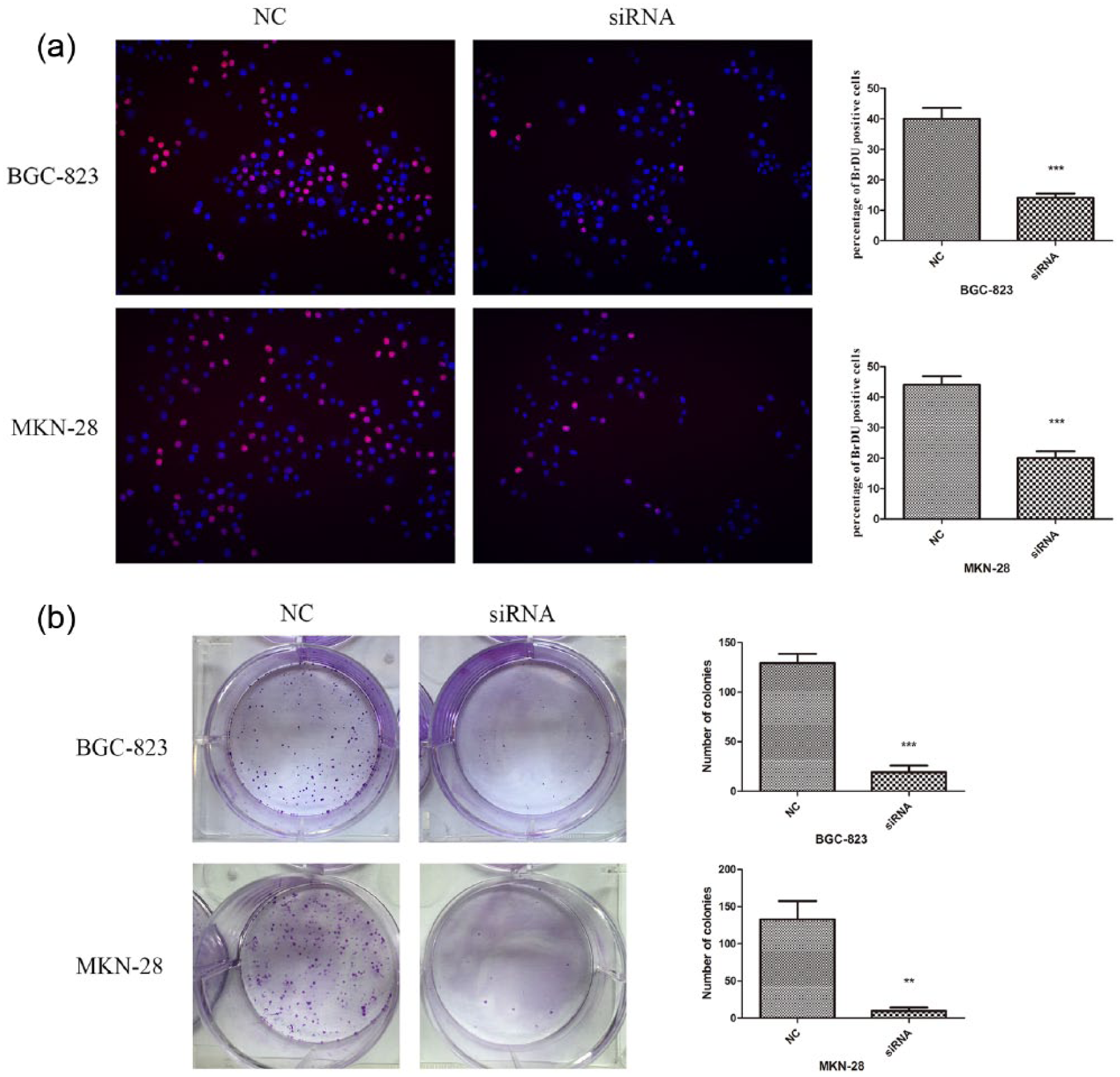
Knockdown of HMGB1 inhibits cell proliferation. (a) Edu-positive cells are shown at 48 h after GC cells (BGC-823 and MKN-28) transfected with HMGB1-siRNA. Original magnification 200×. (b) Colony formation assay. The transfected cells were cultured in six-well plates for 12 days, and the number of colonies was counted and analyzed. The results showed that downregulation of HMGB1 remarkably inhibited the proliferation ability of GC cells. The data are presented as the means ± SD of three independent experiments.
HMGB1 knockdown induces GC cell apoptosis
To investigate the effect of HMGB1 downregulation on cell apoptosis, flow cytometry using Annexin V-FITC/PI double staining was performed. The total apoptosis rates of the HMGB1-siRNA groups were significantly higher than those of the NC groups (BGC-823, p < 0.01; MKN-28, p < 0.05; Figure 4(a)). To further examine the potential molecular mechanisms by which HMGB1 inhibition enhanced cell apoptosis, apoptosis-associated proteins were detected by western blot. The results demonstrated that the expression of Bax was significantly increased and Bcl-2 was markedly decreased in the HMGB1-siRNA groups compared with the NC groups (Figure 4(b)).

Knockdown of HMGB1 induces apoptosis of GC cells. (a) Significant increase in apoptosis in BGC-823 and MKN-28 cells was detected in HMGB1-siRNA–transfected cells. At 48 h after transfection, cell apoptosis was determined using Annexin V-FITC/PI double-staining assay. (b) Western blot showed that HMGB1 knockdown led to upregulation of Bax and downregulation of Bcl-2. The data are presented as the means ± SD of three independent experiments.
HMGB1 knockdown inhibits GC cell migration and invasion
As indicated in Figure 5(a) and (b), in a transwell assay, the ability of cell migration and invasion was significantly inhibited in the HMGB1-siRNA groups compared with that in the NC groups (all p < 0.001). Given that the crucial roles of MMP2 in tumor cell migration and invasion, western blot was performed to measure the expression of this protein. The data revealed that the expression of MMP2 was downregulated in the HMGB1-siRNA groups compared with the NC groups (Figure 5(c)).

Knockdown of HMGB1 inhibits migration and invasion of GC cells. (a) and (b) Transwell migration and invasion assays showed that HMGB1 silencing inhibited migration and invasive ability of BGC-823 and MKN-28 cells, respectively. Original magnification 200×. (c) Western blot indicated that HMGB1 knockdown reduced MMP2 expression. The data are presented as the means ± SD of three independent experiments.
Effects of HMGB1 knockdown on NF-κB/p65
To further explore the potential downstream mechanism of HMGB1-siRNA–mediated biological changes, the expression levels of NF-κB/p65 was analyzed by western blot assay. In addition, the nucleic localization of NF-κB/p65 was detected by immunofluorescence. Results demonstrated that the protein level of NF-κB/p65 was significantly lower in HMGB1-siRNA–transfected cells compared with siNC-transfected cells (BGC-823, p < 0.05; MKN-28, p < 0.01; Figure 6(b)). In addition, downregulation of HMGB1 by HMGB1-siRNA repressed the translocation of NF-κB/p65 from the cytoplasm to the nucleus (Figure 6(a)).

Effects of HMGB1 knockdown on NF-κB/p65. (a) Immunofluorescence showed that HMGB1 downregulation inhibited translocation of NF-κB/p65 from the cytoplasm to the nucleus in BGC-823 and MKN-28 cells, respectively. (b) Western blot detected that HMGB1 knockdown reduced NF-κB/p65 expression. The data are presented as the means ± SD of three independent experiments.
Discussion
In this study, to investigate the role of HMGB1/TLR4/MyD88 signaling in GC and whether it could be used as a potential therapeutic target, we detected the expression of HMGB1, TLR4, and MyD88 in GC specimens and also used RNAi technology to knockdown the HMGB1 gene in GC cell lines. We observed that HMGB1, TLR4, and MyD88 were frequently overexpressed in GC specimens compared with the adjacent non-cancerous tissues, in accordance with the previous studies.11,22,23 Moreover, HMGB1, TLR4, and MyD88 overexpression were shown to be closely related with TNM stage and lymph node metastasis; a high co-expression of HMGB1/TLR4/MyD88 was significantly associated with TNM stage and lymph node metastasis; of these three proteins, a moderately weak correlation with each other was also shown. In addition, we indicated that knockdown of HMGB1 by HMGB1-siRNA suppressed the expression of TLR4 and MyD88, and inhibited cell growth, migration, and invasion in GC cells. Our findings suggest that HMGB1/TLR4/MyD88 signaling may be involved in GC progression.
Previous studies have demonstrated that HMGB1 is overexpressed in many types of cancers.7–11 Furthermore, accumulating evidence has confirmed that HMGB1 is in association with all the hallmarks of cancer, including apoptosis, angiogenesis, invasiveness, metastasis, and inflammatory microenvironment. 24 Interestingly, HMGB1, as a DAMP molecule, is implicated in inflammation, cell differentiation, cell migration, and cancer metastasis by interacting with individual surface receptors, such as RAGE, TLR2, and TLR4.5,6 HMGB1 was indicated to be a prominent synergistic effector for CpG oligonucleotide (CpG ODN) to strengthen the progression of 95D lung cancer cell through MyD88-dependent TLR4 and RAGE signaling. 25 A prominent role for TLRs and innate immune responses in inflammation-related carcinogenesis is demonstrated by a large number of studies. Recent evidence has suggested that TLR4/MyD88 signaling promotes tumor growth in numerous organs. 26 It has been reported that activation of TLR4 signaling contributes to cancer progression by stimulating mitochondrial reactive oxygen species (ROS) production in GC. 22 In addition, it was shown that high expression of MyD88 was associated with tumor progression in hepatocellular carcinoma, and MyD88 silencing decreased tumor growth, invasion and lung metastasis. 27 Recently, TLR4- and MyD88-specific binding has been indicated to play an important role in promoting tumor growth and metastasis in breast cancer 28 and colorectal cancer. 29 Collectively, these data indicate a strong contribution of HMGB1/TLR/MyD88 signaling to tumorigenesis.
Our study revealed that silencing HMGB1 gene by siRNA suppressed cell proliferation and colony formation of GC cells. In addition, we observed that apoptosis was promoted in the HMGB1-siRNA group with significantly elevated expression of Bax and reduced expression of Bcl-2. Bax and Bcl-2 involve in pro-apoptotic and anti-apoptotic signaling, respectively, and are crucial regulators of apoptosis. 30 Moreover, this study showed that downregulation of HMGB1 could inhibit the migration and invasion of GC cells with decreased expression of MMP2, the key enzyme involved in degradation of extracellular matrix (ECM). Wang et al. 25 indicated that HMGB1, interacting with TLR4 and RAGE, could augment the invasive potential of 95D lung cancer cell by upregulating the expression of MMP2 and MMP9 via MyD88-dependent pathway. These results indicate that HMGB1 signaling plays a crucial role in GC development and progression.
Existing evidence has provided that the activity of the NF-κB pathway is prominently involved in the inflammation-associated carcinogenesis and tumor development, 31 and NF-κB has been broadly considered to be overactivated in cancer and, more importantly, to act as the main transcription factor contributing to tumor progression. 32 Sato et al. 33 reported that HMGB1 might continuously activate TLRs, resulting in activation of the NF-κB and MAPK signaling. HMGB1 was revealed to contribute to lung cancer development by activation of Erk1/2 and p38MAPK pathways which lead to the transcriptional regulation of NF-κB. 10 NF-κB upregulates the expression of inflammatory cytokines acting as tumor growth factors for colitis-related cancer. 34 In this study, we demonstrated that downregulated expression of HMGB1 significantly repressed the protein level of NF-κB/p65 and the nuclear translocation of NF-κB/p65 in GC cells, indicating that HMGB1 signaling has a vital function on carcinogenesis and tumor development of GC via the NF-κB signaling pathway.
In summary, our study showed the enhanced HMGB1, TLR4, and MyD88 expression in more aggressive GC tumors. Moreover, blockage of HMGB1/TLR4/MyD88 signaling by HMGB1-siRNA in GC cells could significantly suppress the cell proliferation, migration, and invasion and, in addition, induce apoptosis of GC cells through the NF-κB pathway. These data suggest that HMGB1/TLR4/MyD88 signaling pathway may contribute to the development and progression of GC and it also represents a novel potential therapeutic target for GC.
Footnotes
Acknowledgements
The authors thank international students of Shandong Experimental High School Kexin Cheng and Keyi Cheng for their contributions to language polishing, grammar modification, as well as data acquisition. The authors thank the staff of Qilu Hospital of Shandong University for help with the clinical data. Y.Y. and T.Z. equally contributed to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Fundamental Research Funds of Shandong University No. 2014QLKY10.