
Abstract
DDB1 and CUL4 associated factor 13 (DCAF13) is a protein coding gene located on chromosome 8q22.3, which is a hotspot amplified in various cancers. DCAF13 has been reported to be frequently amplified in breast cancer patients. However, the genetic alteration and potential role of DCAF13 in other cancers, including hepatocellular carcinoma, have not been investigated yet. In this study, we found that DCAF13 was amplified in 14.7% of the cases and its expression was upregulated (p < 0.001) in hepatocellular carcinoma samples in The Cancer Genome Atlas dataset. Increased expression of DCAF13 was also noticed in 40 paired hepatocellular carcinoma and adjacent non-tumor tissues both at messenger RNA and protein levels (p = 0.0002 and 0.0016, respectively). A positive relationship was observed between augmented DCAF13 levels and poorer tumor grade (p = 0.005), and we also found that hepatocellular carcinoma patients with increased DCAF13 expression in their tumors had significantly poorer survival compared with those with decreased DCAF13 expression (median survival time: 45.73 and 70.53 months, respectively). Multivariate Cox regression analysis showed that DCAF13 was an independent prognostic predictor of survival in hepatocellular carcinoma patients. Gene ontology and Kyoto Encyclopedia of Genes and genomes analysis indicated the potential role of DCAF13 as a crucial cell cycle regulator. Collectively, our findings revealed that the overexpression of DCAF13 in hepatocellular carcinoma was significantly associated with poor survival and may participate in the regulation of cell cycle progression.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the second leading cause of cancer-related deaths. More than 700,000 new HCC cases and deaths occur yearly worldwide, with China alone accounting for approximately 50%.1–3 Despite the progress in curative treatments in the management of early-stage HCC, the prognosis of HCC remains poor due to a low rate of early diagnosis and lack of treatment options in advanced stage.4–6 HCC is a complex and heterogeneous tumor with different outcomes and treatment responses even in patients with the same tumor stage. Therefore, identification of molecular biomarkers that accurately predict clinical outcomes could be of substantial aid in better patient stratification and may even lead to novel targeted therapies in specific subsets of patients.7,8
DDB1 and CUL4 associated factor 13 (DCAF13), also known as WD repeat and SOF domain-containing protein 1 (WDSOF1), is a protein coding gene located on chromosome 8q22.3, which is a hotspot amplified in various cancers.9–15 DCAF13 is validated as an RNA binding protein and can be a substrate receptor for CUL4-DDB1 E3 ubiquitin-protein ligase complex.16–18 A genome-wide RNA interference (RNAi) screen identified DCAF13 as a RAS synthetic gene in colon cancer cell lines. 19 DCAF13 was also frequently amplified in breast cancer patients, and the amplification of DCAF13 defined a subgroup of breast cancer patients with significant worse prognosis. 13 Collectively, the findings above indicate that DCAF13 might play a significant role in cancer biology and serve as an ideal biomarker for better stratification of cancer patients.
In this study, we investigated the expression and clinical significance of DCAF13 in HCC. Moreover, we predicted the possible biological processes regulated by DCAF13 using bioinformatics methods.
Materials and methods
Data mining and The Cancer Genome Atlas database analysis
The Cancer Genome Atlas (TCGA) database stores genomic and clinical data of a series of cancers including HCC, and the data have been made public for appropriate analysis according to the publication guideline. 20 The gene expression (normalized RNA-seq by expectation maximization value, log2 transformed) and clinical data of HCC patients were downloaded from the TCGA data portal. Then, 360 pathological confirmed primary HCC and 49 corresponding adjacent non-tumor samples were selected for further analysis (3 fibrolamellar carcinoma and 7 hepatocholangiocarcinoma samples were excluded). Genetic alterations, including copy number status and mutation of the selected TCGA HCC samples, were retrieved in the cBioPortal platform.21,22
Another RNA sequencing (RNA-seq) dataset comprising 50 HCC samples (GSE65485) was downloaded from Gene Expression Omnibus (GEO) datasets, and the processed expression value (FPKM (fragments per kilobase million)) was used in our study. 23
Patients, samples, and cell lines
A total of 360 primary HCC samples and 49 adjacent non-tumor controls from the TCGA dataset (TCGA cohort) were included in this study. Age, gender, tumor grade, tumor–node–metastasis (TNM) stage, alpha-fetoprotein (AFP) value, vascular invasion, overall survival, and corresponding vital status were evaluated and are shown in Table 1. Overall survival was defined as the time from the date of surgery to the date of death or last follow-up. Follow-up was completed on 28 January 2016.
Patients’ clinicopathological characteristics and DCAF13 expression.

DCAF13: DDB1 and CUL4 associated factor 13; TNM: tumor–node–metastasis; AFP: alpha-fetoprotein.
p < 0.01; ***p < 0.001.
A total of 40 pairs of fresh HCC and adjacent non-tumor samples were obtained from patients who underwent hepatectomy for HCC in Peking Union Medical College Hospital, Beijing, China. Samples were stored in liquid nitrogen. The study protocol was approved by the Ethics Committee of Peking Union Medical College Hospital. Informed consent was obtained from each patient.
The immortalized human hepatocyte (IHH) cell line was a kind gift from Dr Chen Deng (Chinese Academy of Medical Science, China). HCC cell lines (HepG2, Hep3B, Bel-7402, and SMMC-7721) were purchased from the Shanghai Cell Bank, Chinese Academy of Sciences. All the cell lines were cultured under standard conditions.
RNA isolation, reverse transcription, and quantitative real-time polymerase chain reaction
Total RNA was extracted from tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed using K1622 RevertAid™ First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using Bio-Rad CFX-96 (Bio-Rad Laboratories, Hercules, CA, USA), and the data were normalized to β-actin messenger RNA (mRNA) expression. The primers for real-time PCR were as follows: DCAF13—F: CTGTTGGAACCCTATGGAA and R: AGATTTATCGAAACTAGCAGAC and β-actin—F: AGAGGGAAATCGTGCGTGAC and R: CAATAGTGATGACCTGGCCGT.
Western blot analysis
The total protein lysate was extracted from HCC and adjacent non-tumor tissues using cell lysis buffer for Western blotting and immunoprecipitation (Beyotime, Shanghai, China). Protein lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 10% separation gel) and transferred onto a polyvinylidene fluoride (PVDF) membrane for immunoblotting. The membranes were blocked with 5% fat-free milk for 1 h at room temperature and then incubated with the following primary antibodies: rabbit anti-human DCAF13 monoclonal antibody (ab195121; 1:5000; Abcam, Cambridge, MA, USA), rabbit anti-human MYC monoclonal antibody (ab32072; 1:0000; Abcam), and mouse anti-human GAPDH monoclonal antibody (CW0100M; 1:5000; ComWin, Beijing, China). Horseradish peroxidase–conjugated secondary antibodies were used. Signals were detected using an enhanced chemiluminescence (ECL) kit (Millipore, Billerica, MA, USA). The integrated option density (IOD) of the protein bands was detected with a Gel-Pro analyzer 32, and the relative protein expression was calculated by normalizing the specific protein IOD value to the corresponding IOD of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Functional enrichment analysis
The Pearson correlation coefficient of the expression of DCAF13 and other genes was calculated using TCGA HCC RNA-seq data. According to the correlation coefficient (r ≥ 0.35), we defined the co-expressed genes with DCAF13 in HCC. The functional enrichment analysis of co-expressed genes was performed to predict the function of DCAF13 using DAVID Bioinformatics Tool.24,25 The enrichment results were reported and limited to gene ontology (GO) terms in the “Biological Process” (GOTERM-BP-FAT) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway categories using the functional annotation clustering. The GO terms and KEGG pathways with p < 0.01 were identified as significantly enriched functional annotations. The heatmap was made in R 3.3.2 using pheatmap.
Statistical analysis
Student’s t test was applied for the comparison of two groups. A one-way analysis of variance (ANOVA) was performed to compare more than two groups. A Pearson chi-square test was used to assess the correlation of DCAF13 expression with the clinicopathological characteristics of HCC patients. The correlation coefficients were determined using the Pearson correlation test. Survival curves were determined using the Kaplan–Meier method and compared using the log-rank test. Hazard ratios (HRs) of death associated with DCAF13 expression and other predictor variables were estimated by univariate Cox proportional hazards regression model and a multivariate Cox model was constructed to estimate the adjusted HR for DCAF13 expression. p < 0.05 were taken as statistically significant. All statistical analyses were conducted using the SPSS statistical software package (version 22.0; IBM, Armonk, NY, USA) and GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
Results
DCAF13 is amplified and upregulated in HCC
We first investigated the genetic alterations of DCAF13 in 360 pathological confirmed HCC in the TCGA dataset using the cBioPortal platform.21,22 Genetic alterations of DCAF13 were found in 15.8% of all cases, and amplification was the predominant type, accounting for 94.7% of all alterations (Figure 1(a)). Plotting of the corresponding mRNA level in relation to copy number status of DCAF13 revealed that the amplification of DCAF13 was associated with a significant increase in mRNA expression (Figure 1(b), p < 0.0001).

DCAF13 is amplified and upregulated in HCC. (a) Genetic alterations of DCAF13 in the TCGA HCC cohort were explored using the cBioPortal website. (b) DCAF13 mRNA expression was significantly associated with the copy number alteration status (ANOVA, p < 0.0001). (c) Analysis of DCAF13 mRNA expression in TCGA HCC samples compared with adjacent non-tumor controls (unpaired t test, p < 0.0001).
Next, we studied the relative expression of DCAF13 in 360 primary HCC tissues versus 49 adjacent non-tumor tissues in the TCGA dataset. The DCAF13 expression level was significantly upregulated in HCC tissues in comparison with the non-tumor controls (Figure 1(c), p < 0.0001).
The mRNA and protein levels of DCAF13 are increased in HCC
A total of 40 pairs of fresh HCC tissues and adjacent non-tumor tissues were used to validate the overexpression of DCAF13 in HCC. First, we measured the DCAF13 mRNA level using real-time PCR. Compared with the corresponding non-tumor tissues, DCAF13 was significantly overexpressed (more than 1.5-fold, log 1.5 [T/N] > 1) in 62.5% of all HCC cases (Figure 2(a), p = 0.0002). Then, we performed Western blot analysis to validate the overexpression of DCAF13 at the protein level. Consistent with the expression of mRNA level, DCAF13 was also upregulated in the majority of the HCC tissues at the protein level (Figure 2(b), p = 0.0016). The representative Western blot images of 20 pairs of HCC tissues and adjacent non-tumor tissues are shown in Figure 2(c).
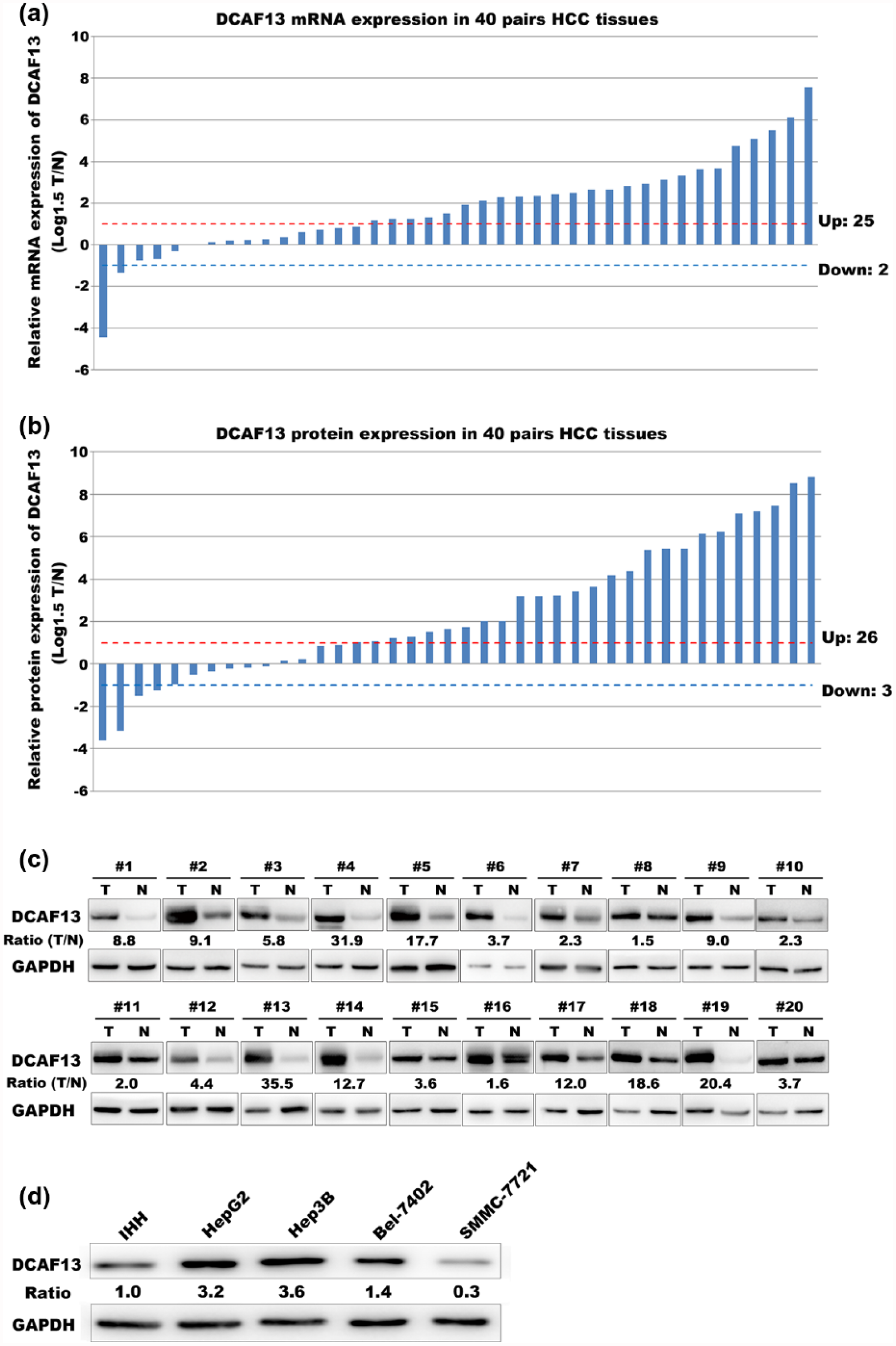
The mRNA and protein levels of DCAF13 are increased in HCC. (a) mRNA expression level of DCAF13 in 40 pairs of HCC and adjacent non-tumor controls (paired t test, p = 0.0002). (b) Protein level expression of DCAF13 in 40 pairs of HCC and adjacent non-tumor controls (paired t test, p = 0.0016). (c) Representative Western blot images of 20 pairs of HCC tissues and adjacent non-tumor tissues. (d) The cell lysates of IHH cell line and four HCC cell lines (HepG2, Hep3B, Bel-7402, and SMMC-7721) were collected. Western blot analysis was performed to detect the protein levels of DCAF13 in these cells using the corresponding antibodies.
Moreover, we compared the DCAF13 expression of four HCC cell lines to that of a normal hepatocyte cell line using Western blot analysis. Consistently, DCAF13 was overexpressed in most of the HCC cell lines (Figure 2(d)). Collectively, these results indicated that DCAF13 was overexpressed in HCC.
Augmented DCAF13 expression correlates with poorer histological grade in HCC patients
We explored the correlation of DCAF13 expression with the clinicopathological features of HCC using the RNA-seq and clinical data of 360 primary HCC patients from the TCGA dataset. As shown in Table 1, a high DCAF13 expression was positively correlated with male sex (p < 0.001) and poorer histological grade (p = 0.005). No significant association was found between DCAF13 expression and age, TNM stage, AFP value, and vascular invasion.
Overexpression of DCAF13 identifies a subgroup of HCC patients with poor survival
Kaplan–Meier analysis showed that HCC patients with DCAF13 overexpression had significantly poorer survival compared with those with low DCAF13 expression (median survival time: 45.73 and 70.53 months, respectively), and the result was statistically significant (p = 0.0036, log-rank test, Figure 3). In the univariate Cox proportional hazard regression analysis, overexpressed DCAF13 was also significantly associated with an increased risk of cancer-related death (HR = 1.691, p = 0.004, Table 2). After adjusting for potential confounding factors, multivariate Cox regression analysis showed that overexpressed DCAF13 was an independent predictor of poorer survival of HCC patients (HR = 2.220, p = 0.003, Table 2).

Overexpression of DCAF13 identified a subgroup of HCC patients with poor survival. Kaplan–Meier analysis showed that DCAF13 overexpression was significantly associated with poorer survival in HCC patients. The difference is statistically significant based on the log-rank test (p = 0.0036).
Cox regression analysis of potential prognostic predictors for HCC patients.

HCC: hepatocellular carcinoma; HR: hazard ratio; CI: confidence interval; TNM: tumor–node–metastasis; AFP: alpha-fetoprotein; DCAF13: DDB1 and CUL4 associated factor 13.
p < 0.05; **p < 0.01; ***p < 0.001.
DCAF13 is a potential regulator of cell cycle progression
We performed bioinformatics analysis to predict the potential function of DCAF13 in HCC. We first calculated the Pearson correlation coefficient between DCAF13 and other genes in RNA-seq data of the TCGA HCC cohort. According to the Pearson correlation coefficient (r ≥ 0.35), we identified 773 genes whose expression followed a similar pattern to that of DCAF13. GO analysis showed that these genes co-expressed with DCAF13 were significantly enriched in 79 GO terms, which were mainly involved in eight functional clusters (p < 0.01, Table 3). As shown in Table 3, the top enriched functional clusters included cell cycle and related process, which indicated the potential association of DCAF13 with cell cycle regulation in HCC tumorigenesis. We further performed KEGG pathway analysis for the co-expressed genes and consistently found that cell cycle pathway was also significantly enriched (Figure 4(a)). Hierarchical clustering analysis was performed for DCAF13 and the enriched cell cycle pathway genes in two different HCC RNA-seq datasets. The results of the clustering analysis suggested that these cell cycle genes may represent a functional module and involve in the DCAF13-regulated program (Figure 4(b) and (c)).
Top five enriched functional clusters of GO terms.

GO: gene ontology; ncRNA: non-coding RNA; rRNA: ribosomal RNA; mRNA: messenger RNA; snRNP: small nuclear ribonucleo protein; tRNA: transfer RNA.

DCAF13 is a potential regulator of cell cycle progression. (a) KEGG pathway analysis of the genes co-expressed with DCAF13 in HCC. (b and c) Heatmap of DCAF13 and the 13 enriched cell cycle pathway genes in the TCGA HCC cohort (n = 360) and GSE65485 (n = 50). Clustering was performed with the hclust function. (d and e) Correlation of MYC mRNA expression with that of DCAF13 in the TCGA cohort and GSE65485. (f) Western blot analysis of DCAF13 and MYC in 24 fresh HCC samples. (g) The correlation between DCAF13 and MYC expression at the protein level in 24 fresh HCC samples (Pearson correlation, r = 0.5824, p = 0.0028).
MYC was a well-established nodal gene in the cell cycle pathway, and the mRNA expression of MYC was significantly correlated with that of DCAF13 (Figure 4(d) and (e)). To further validate the co-expression of DCAF13 and MYC expression at the protein level, we performed Western blot analysis of 24 HCC tissue samples. As a result, the correlation of DCAF13 expression with MYC expression was also verified at the protein level (Figure 4(f) and (g), r = 0.5824, p = 0.0028).
Discussion
DCAF13, also known as WDSOF1, is located at locus 8q22.3, which is a hotspot for amplification in various cancers.9–15 Chin et al. 13 reported that DCAF13 (WDSOF1) was one of the most frequently amplified genes in breast cancer and showed coordinate overexpression. Consistently, we found that DCAF13 was also amplified frequently (15.8%) in HCC patients in a TCGA dataset. Then, we found that DCAF13 was upregulated in HCC and that its amplification may contribute, at least partly, to its upregulation. In addition, we then validated DCAF13 overexpression not only at the mRNA level but also at the protein level using 40 pairs of HCC and adjacent non-tumor tissues.
Chin et al. also reported that the amplification of DCAF13 (WDSOF1) significantly correlated with worse overall survival in breast cancer patients. 13 In our study, we found that HCC patients with DCAF13 amplification had poorer overall survival than those without DCAF13 amplification (45.5 vs 58.8 months), but the difference did not achieve statistical significance (p = 0.196). Nevertheless, we found a significant association between the expression of DCAF13 and the overall survival of HCC patients using Kaplan–Meier analysis. Multivariate Cox regression analysis further determined the overexpressed DCAF13 as an independent predictor for poorer overall survival. As to clinicopathological characteristics, we found that DCAF13 overexpression significantly correlated with male sex and a poorer tumor grade. The correlation of DCAF13 overexpression with the AFP value and vascular invasion was marginally significant.
Luo et al. 19 carried out a genome-wide RNAi screen in colorectal cancer cell line and found that DCAF13 (WDSOF1) exhibited synthetic lethal interaction with the KRAS oncogene. To explore the potential function of DCAF13 in HCC tumorigenesis, we performed bioinformatics analysis using genes co-expressed with DCAF13. GO biological process analysis showed eight enriched functional clusters, more than half of which associated with RNA metabolism (mRNA processing, ribosome biogenesis, transfer RNA (tRNA) metabolic process, RNA transport, and posttranscriptional regulation of gene expression), and this was indeed consistent with the proved role of DCAF13 as an RNA binding protein.16,17 The enriched functional clusters also included the regulation of protein ubiquitination, which was in line with the reported function of DCAF13 as a substrate receptor for CUL4-DDB1 E3 ubiquitin-protein ligase complex. 18 The above findings supported the appropriate use of the functional enrichment analysis of the co-expressed genes to predict the potential function of DCAF13.26,27 Therefore, the finding that cell cycle and related process were also significantly enriched made us to consider the potential role of DCAF13 in cell cycle regulation in HCC tumorigenesis. Consistently, further KEGG pathway analysis demonstrated the significant enrichment of the cell cycle pathway. Of the 13 enriched cell cycle pathway genes, MYC is the most famous and important oncogene involved in cell cycle regulation.28–30 In our study, we found that there was a significant positive correlation between DCAF13 and MYC mRNA expression in two different HCC RNA-seq datasets. To further confirm the co-expression of DCAF13 and MYC at the protein level, we performed Western blot of fresh HCC samples and validated the significant co-expression relationship between DCAF13 and MYC at the protein level. These findings indicated that DCAF13 might be an oncogene involved in the MYC pathway that regulates cell cycle progression.
In conclusion, we discovered and validated the upregulation of DCAF13 in HCC. Our findings suggested that DCAF13 might serve as a novel prognostic biomarker and a potential therapeutic target in HCC. However, further research is required to clarify the exact biological function and molecular mechanism of DCAF13 in HCC.
Footnotes
Acknowledgements
The authors thank Qiaofei Liu from Peking Union Medical College Hospital (PUMCH) for technical assistance. Jia.C., P.H., and Jie.C. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (91129716 to X.D.H.).