
Abstract
The major cause of death in osteosarcoma is the invasion and metastasis. Better understanding of the molecular mechanism of osteosarcoma invasion is essential in developing effective tumor-suppressive therapies. Interaction between chemokine receptors plays a crucial role in regulating osteosarcoma invasion. Here, we investigated the relationship between CXCR7 and CXCR4 in osteosarcoma invasion induced by bone marrow microenvironment. Human bone marrow mesenchymal stem cells were co-cultured with osteosarcoma cells to mimic actual bone marrow microenvironment. Osteosarcoma cell invasion and CXCL12/CXCR4 activation were observed within this co-culture model. Interestingly, in this co-culture model, osteosarcoma cell invasion was not inhibited by suppressing CXCR4 expression with neutralizing antibody or specific inhibitor AMD3100. Downstream signaling extracellular signal–regulated kinase and signal transducer and activator of transcription 3 were not significantly affected by CXCR4 inhibition. However, suppressing CXCR4 led to CXCR7 upregulation. Constitutive expression of CXCR7 could maintain osteosarcoma cell invasion when CXCR4 was suppressed. Simultaneously, inhibiting CXCR4 and CXCR7 compromised osteosarcoma invasion in co-culture system and suppressed extracellular signal–regulated kinase and signal transducer and activator of transcription 3 signals. Moreover, bone marrow microenvironment, not CXCL12 alone, is required for CXCR7 activation after CXCR4 suppression. Taken together, suppressing CXCR4 is not enough to impede osteosarcoma invasion in bone marrow microenvironment since CXCR7 is activated to sustain invasion. Therefore, inhibiting both CXCR4 and CXCR7 could be a promising strategy in controlling osteosarcoma invasion.
Introduction
Osteosarcoma (OS) is the most common primary malignant bone tumor, mainly affecting children and young adults. 1 The past few decades have witnessed profound advances in diagnosis and treatment of OS. Though modern chemotherapy in conjunction with surgery achieves a 5-year event-free survival of 60%–70% in extremity localized, non-metastatic disease, a major problem is the poor prognosis for metastatic relapse or recurrence. 2
The interactions between bone marrow mesenchymal stem cells (BMSCs) and malignant tumors from the breast, 3 prostate, 4 kidney, 5 and pancreas6,7 have been recently described. However, the mechanism of such interaction was seldomly reported. Here, we used BMSCs co-culture model to mimic bone marrow (BM) microenvironment and investigated its effect on OS cells. Chemokine CXCL12, secreted by BMSCs, and its receptor CXCR4 were involved in invasion and metastasis of OS. 8 Moreover, CXCL12/CXCR4 activation was found to be correlated with poor overall survival of OS patients. 9 However, there were contradictory discussions about the function of CXCR4 in OS. Ma et al. 10 found no evidence that CXCR4 expression was related to invasion and metastasis activity in primary tumor tissue. Moreover, Baumhoer et al.’s 11 report suggested that a favorable outcome in OS is associated with CXCL12/CXCR4 activation. In OS, the function of CXCL12/CXCR4 axis is still awaiting further elucidation.
CXCR4 has long been considered as the only receptor that binds to CXCL12. However, CXCR7 was recently discovered as a second receptor with high affinity for CXCL12. 12 The majority of the reports drew the conclusion that CXCR7 serves as a positive regulator in enhancing proliferation/metastasis of tumors.13–16 Similar to CXCR4, the function of CXCR7 in tumor cells is still controversial. It was revealed by Liberman et al. 17 and Uto-Konomi et al. 18 that CXCR7 acted as a negative regulator for CXCL12/CXCR4 axis and elicited anti-tumorigenic effects.
In this study, we explored the interaction between CXCR4 and CXCR7 in OS cells. BMSCs co-culture environment activated CXCR4 and promoted invasion of OS cells. OS cells invasion was not significantly affected by CXCR4 suppression within this co-culture model. Interestingly, CXCR7 expression was elevated when CXCR4 was suppressed. Elevated CXCR7 expression was responsible for the sustained OS invasion in BM microenvironment, and dual inhibition of CXCR4 and CXCR7 achieved a significant inhibition on OS invasion. These results shed light on the functions of chemokine receptors and proposed a new strategy in controlling OS invasion.
Materials and methods
Cells and reagents
Human OS cell lines MG-63 and U2OS were purchased from the Chinese Academy of Sciences (Shanghai, China). OS cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM). Immortalized Human Bone Marrow Mesenchymal Stem Cells SV40 (T0520) was purchased from abm (Richmond, British Columbia, Canada) and maintained in Prigrow III medium (ABM Inc, Cat. No. TM003). To make the complete growth medium, the following components were added to the base medium: fetal bovine serum (FBS, Gibco, Grand Island, NY) to a final concentration of 10%, 100 U/mL penicillin G, and 100 U/mL streptomycin. Recombinant human CXCL12 (Stromal cell–derived factor 1 (SDF-1)) and CXCR4 neutralizing antibody 12G5 (MAB170) was brought from R&D Systems (Minneapolis, MN). CXCR4 antagonist AMD3100 and Matrigel were obtained from Sigma-Aldrich (St. Louis, MO). The inhibitors of endosomal and lysosomal protein degradation, bafilomycin A1 (B1793) and chloroquine diphosphate (C6628), were purchased from Sigma-Aldrich (Deisenhofen, Germany). In western blot assay, CXCR4 (GTX31630) and CXCR7 (GTX100027) antibodies were brought from GeneTex (Irvine, CA) and used at 1:1000 dilution. Anti-p-ERK1/2 (pT202/pY204.22A; sc-136521) and pY705-Stat3 (sc-7993) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and used at 1:500 dilution. MMP9 (13667) and MMP2 (13132) antibodies were purchased from Cell Signaling Technology (Danvers, MA) and used at 1:1000 dilution. The antibody of β-actin (BM0005, 1:2000 dilution) was purchased from Boster (Wuhan, China).
Transwell co-culture system
BMSCs were cultured in apical compartments of transwells (transwell insert 0.4 µm; Millipore, Billerica, MD) with OS cells grown in the basal compartment of the plate (EMD Millipore). BMSCs were seeded onto the upper layer of transwells. OS cells were seeded onto the lower layer of transwells. After different treatments, OS cells were collected at indicated time for further analysis.
Invasion assay
Invasion assay was performed using Boyden chambers with inserts (pore size: 8 µm) coated with Matrigel in 24-well plates. Briefly, after different treatments, MG-63 or U2OS cells (4 × 105 cells/mL) were suspended in 250 µL medium in the upper chamber, which was pre-coated with 15 µL Matrigel (0.5 µg/mL). BMSCs were seeded onto the lower layer of transwells (2 × 105 cells/well). The control group consisted of plates with OS cells in the upper chamber only. The plates were incubated for 72 h at 37°C. Invaded cells on the lower surface were fixed with 100% methanol and stained with hematoxylin and eosin. For each group, the invaded cells in five randomly chosen fields were quantified by manual counting.
ELISA assay
After culturing BMSCs, MG-63, and U2OS cells for 72 h, the medium was collected separately and centrifuged at 1000 r/min for 5 min. The supernatants were then stored at −80°C for further enzyme-linked immunosorbent assay (ELISA) test. ELISA was performed using the human CXCL12/SDF-1 kit (R&D Systems) according to the manufacturer’s instructions.
Western blot analysis
After different treatments, OS cells were harvested and lysed in lysis buffer. The mixture was centrifuged at 14,000g at 4°C for 30 min, and a bicinchoninic acid assay (BCA) assay was performed to detect the concentration of protein in the supernatants using a Varioskan multimode microplate spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Proteins extracts were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were then transferred by semi-dry blotting to nitrocellulose (NC) membranes. The NC membranes were blocked with 10% no-fat milk in phosphate-buffered saline (PBS) at 37°C for 1 h and incubated with specific antibodies against indicated proteins overnight at 4°C. After washing three times for 10 min each in phosphate-buffered saline with Tween 20 (PBST), the membranes were incubated with IRDyeTM800-conjugated secondary antibody (1:1000, LI-COR) for 1 h at 37°C. Detection was performed using the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
RNA isolation and reverse transcription polymerase chain reaction
MG-63 or U2OS cells were treated with AMD3100 or neutralizing antibody, then total messenger RNA (mRNA) was isolated using TRIzol reagent (Invitrogen, Calsbad, CA, USA) according to the manufacturer’s instructions. Reverse transcription (RT) was performed using equal amounts of mRNA and the real-time quantitative polymerase chain reaction (PCR) was performed using the Takara kit (Takara Bio Inc., Otsu, Shiga, Japan); complementary DNA was collected and saved for real-time quantitative RT-PCR (qRT-PCR). qRT-PCR was performed in a Chromo4 instrument (Bio-Rad, Hercules, California) with the SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA). The primer sequences are as follows: CXCR4: 5′-CAAGGGCCACCAGAAGCGCAA-3′ and 3′-TTTGGAGAGGATCTTGAGGCTGGA-5′ and for glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 5′-CATCTTCCAGGAGCGAGATCC-3′ and 3′-GGTGCAGGTGGCATTGCTGATG-5′.
Gelatin zymography
MMP2 and MMP9 activities were assessed by gelatin zymography. MG-63 and U2OS cells were cultured (1 × 106 cells/mL) for different treatments. Samples were prepared in non-reducing conditions in 5× sample buffer (0.625 M Tris-HCl, 10% glycerol, 2% SDS, 2% Bromophenol blue) and resolved on 8% SDS-PAGE containing 0.5 mg/mL of gelatin Type A from porcine skin (Sigma-Aldrich, St. Louis, MO). The gels were then washed with 2.5% (v/v) Triton X-100 for 10 min at room temperature for three times. Then, the gels were incubated in developing buffer (100 mM Tris-HCl, pH 7.9, 30 mM CaCl2, and 0.02% sodium azide) for 30 min at room temperature followed by a second incubation in fresh developing buffer overnight at 37°C. The gels were then stained with Simply Blue Safe Stain solution (Life Technologies, Grand Island, NY) for 15 min, followed by de-staining in water to reveal the areas of activity. 19
Stable silencing of CXCR7 by short hairpin RNA
CXCR7 short hairpin RNA (shRNA) and scrambled sequence shRNA (control shRNA) constructs were cloned into pRS plasmid under the control of U6 promoter for stable expression (HuSh-29mer, OriGene, Rockville, MD). CXCR7-shRNA primer sequence is 5′-GCATCTCTTCGACTACTCAGA-3′. The scrambled shRNA was used as a negative control (referred to as shControl in the text), whose sequence is 5′-GACGAGCTTCTACACAATCAT-3′. MG-62 and U2OS cells were transfected with shRNA-pRS using Lipofectamine 2000 (Invitrogen). Stable transfectants were selected in puromycin selection medium (2.0 mg/mL) for 2 weeks. Then, the emergent cell colonies were evaluated for CXCR7 expression by western blot analysis.
Stable expression of CXCR7 in MG-63 and U2OS cells
MG-63 and U2OS cells were transfected with a human full-length CXCR7cDNA (True Clones, pCMV6-Neo vector; OriGene) using Lipofectamine 2000. Stable clones were selected with G418 (250 mg/mL) and analyzed for CXCR7 expression by immunoblotting. Cell colonies overexpressing CXCR7 were pooled for further analysis.
Statistical analysis
All data in the experiments were expressed as mean ± standard error of the mean (SEM). Data were obtained from at least three independent experiments. Statistical analyses were performed using an unpaired, two-tailed Student’s t test.
Results
CXCL12/CXCR4 activation directs OS invasion in BM microenvironment
Human BMSCs co-culture model was used to simulate BM microenvironment. Within 72 h of BMSCs co-culture, increased invasion was observed in both MG-63 and U2OS cells (Figure 1(a) and (b)). ELISA assay was used to determine the amount of CXCL12 secreted by BMSCs, MG-63, and U2OS cells. ELISA results proved that BMSCs, but not OS cells, secreted CXCL12 (Figure 1(c)). Furthermore, CXCR4 expression was detected with western blot in OS cells. BMSCs co-culture elevated CXCR4 expression in both MG-63 and U2OS cells (Figure 1(d) and (e)). Therefore, BMSCs co-culture induced OS invasion through activating CXCL12/CXCR4 axis.
Effect of CXCR4 suppression on OS cell migration
CXCR4 suppression was found to be effective in inhibiting cancer invasion and metastasis. 20 MG-63 and U2OS cells were treated with CXCR-4-neutralizing antibody (12G5; 10 µg/mL) or CXCR4-specific chemical inhibitor AMD3100 (500 ng/mL); CXCR4 expression in MG-63 (Figure 2(a) and (b)) and U2OS (Figure 2(c) and (d)) cells was significantly reduced. Result of RT-PCR indicated that CXCR4 mRNA was not affected by those treatments (Figure 2(e)). Further investigation showed that AMD3100 treatment results in CXCR4 degradation via endosomal–lysosomal proteolysis. We delivered endosomal and lysosomal proteolysis inhibitors bafilomycin A1 (100 mM) or chloroquine (200 mM) 2 h prior to AMD3100 treatment. Western blot results revealed that inhibition of endosomal–lysosomal proteolysis restored CXCR4 levels which were reduced by AMD3100 (Figure 2(f)–(i)). However, OS cell invasion was not significantly affected by AMD3100 or CXCR4 antibody treatment within 72 h of BMSCs co-culture (Figure 2(j) and (k)).
CXCR7 signal was activated after CXCR4 inhibition in OS cells
The influence of BMSCs on OS cells was further investigated by testing the activation of chemokine receptor signals. After AMD3100 and neutralizing antibody treatment, MG-63 and U2OS showed decreased expression of CXCR4 and elevated expression of CXCR7 (Figure 3(a)–(c)). Expression of phosphorylated signal transducer and activator of transcription 3 (p-STAT3) and extracellular signal–regulated protein kinases 1 and 2 (p-ERK1/2) remained unchanged in both the cells after CXCR4 inhibition within BMSCs co-culture environment (Figure 3(d)–(f)), suggesting that STAT3 and ERK signal activities were not affected by CXCR4 inhibition. MMP9 and MMP2 were identified as predictive markers of OS cell invasion. 21 Therefore, we assessed the expression of MMP9 and MMP2 in MG-63 and U2OS cells. The results showed that MMP9 and MMP2 protein levels in both the cells were upregulated in the co-culture system and were not affected by CXCR4 inhibition (Figure 3(g)–(i)). Furthermore, consistent with an elevation in MMP9 and MMP2 protein expression, an increase in MMP9 (Figure 3(j)) and MMP2 (Figure 3(k)) enzymatic activity was also observed using gelatin zymography, demonstrating the functional consequences that result from increased protein expression. These results suggested that in BM microenvironment, activation of CXCR7 might be responsible for OS cell invasion when CXCR4 was inhibited.
Constitutive expression of CXCR7 restored OS cell invasion which was inhibited by CXCR4 suppression
To further confirm the effect of CXCR7 on OS invasion, MG-63 and U2OS cells cultured in regular medium were transfected with CXCR7. The CXCR7-transfected OS cells showed an increase in CXCR7 expression (Figure 4(a) and (b)). Furthermore, CXCR4 inhibition compromised OS cell invasion in regular medium. Elevated CXCR7 expression maintained invasion when CXCR4 was inhibited in both the cell lines (Figure 4(c) and (d)). High level of CXCR7 also restored MMP9 and MMP2 expression, which were downregulated by CXCR4 inhibition, in MG-63 and U2OS cells (Figure 4(e)–(g)). This further confirmed that switching from CXCR4 to CXCR7 contributed to the maintenance of OS cell invasion.
Dual inhibition of CXCR4 and CXCR7 showed a strong effect against OS cell invasion
CXCR7-shRNA transfection was verified by testing CXCR7 protein expression with western blot (Figure 5(a) and (b)). In BMSCs co-culture environment, invasion of MG-63 (Figure 5(c)) and U2OS (Figure 5(d)) was significantly inhibited when cells were treated with combination of CXCR7-shRNA and CXCR4 inhibition (AMD3100 or CXCR4 neutralizing antibody). Dual inhibition on CXCR7 and CXCR4 expression was further confirmed by western blot (Figure 5(e)–(g)). Expression of downstream molecules, p-STAT3 and p-ERK1/2, was reduced by combined treatment in both OS cell lines (Figure 5(h)–(k)). The inhibitory effect on OS invasion was further verified by testing expression of MMP9 and MMP2. Western blot assay showed significant inhibitory effect on MMP9 and MMP2 expression when CXCR4 and CXCR7 were both suppressed (Figure 5(h)–(k)).
BM microenvironment is required for switch from CXCR4 to CXCR7 in OS cells
Since CXCL12 was proved to be one of the key factors that induce cancer invasion 22 and BMSCs secreted a high level of CXCL12 (Figure 2(c)), we added CXCL12 (100 ng/mL) into the medium to investigate the major reason for cell signal switching in OS cells. However, simply adding CXCL12 into the medium did not replicate the signal switching which happened in BMSCs co-culture model (Figure 6(a)–(c)). Moreover, invasion assay showed that CXCL12 did not sustain OS cell invasion when cells were treated with AMD3100 or CXCR4 neutralizing antibody (Figure 6(d) and (e)). Reduced MMP9 and MMP2 expression also indicated the inhibitory effect of CXCR4 inhibition on OS cell invasion (Figure 6(f)–(h)). CXCL12 alone did not trigger cell signal switch or sustain OS cell invasion after CXCR4 inhibition. These results suggested that BM microenvironment is required for the sustainment of OS cell invasion when CXCR4 was inhibited.

CXCL12/CXCR4 activation directs OS invasion in bone marrow microenvironment. (a and b) MG-63 and U2OS cells were incubated with BMSCs for 72 h, then cell invasion was measured with the Transwell. (c) CXCL12 levels in supernatants of BMSCs, MG-63, and U2OS cells were detected using ELISA. (d and e) CXCR4 protein expression in MG-63 and U2OS cells was tested using western blot. RM stands for regular medium and CM stands for co-culture medium. Data were mean ± SD for three independent experiments (**p < 0.01 compared with BMSCs group or regular medium groups).

Effect of CXCR4 suppression on OS cell invasion. MG-63 and U2OS cells were treated with AMD3100 or CXCR4 neutralizing antibody and co-cultured with BMSCs for 72 h. Effect of AMD3100 or CXCR4 neutralizing antibody on CXCR4 expression in (a and b) MG-63 and (c and d) U2OS cells was verified by western blot. (e) CXCR4 mRNA level was measured by RT-PCR. MG-63 and U2OS cells were treated with endosomal and lysosomal proteolysis inhibitors bafilomycin A1 (100 mM) or chloroquine (200 mM) 2 h prior to AMD3100 treatment. CXCR4 expression in (f and g) MG-63 and (h and i) U2OS cells were analyzed by western blot. Invasion of (j) MG-63 and (k) U2OS cells were measured with the Transwell. CM stands for co-culture medium. Data were mean ± SD for three independent experiments (**p < 0.01 compared with control or regular medium groups; ##p < 0.01 compared with AMD3100 groups).
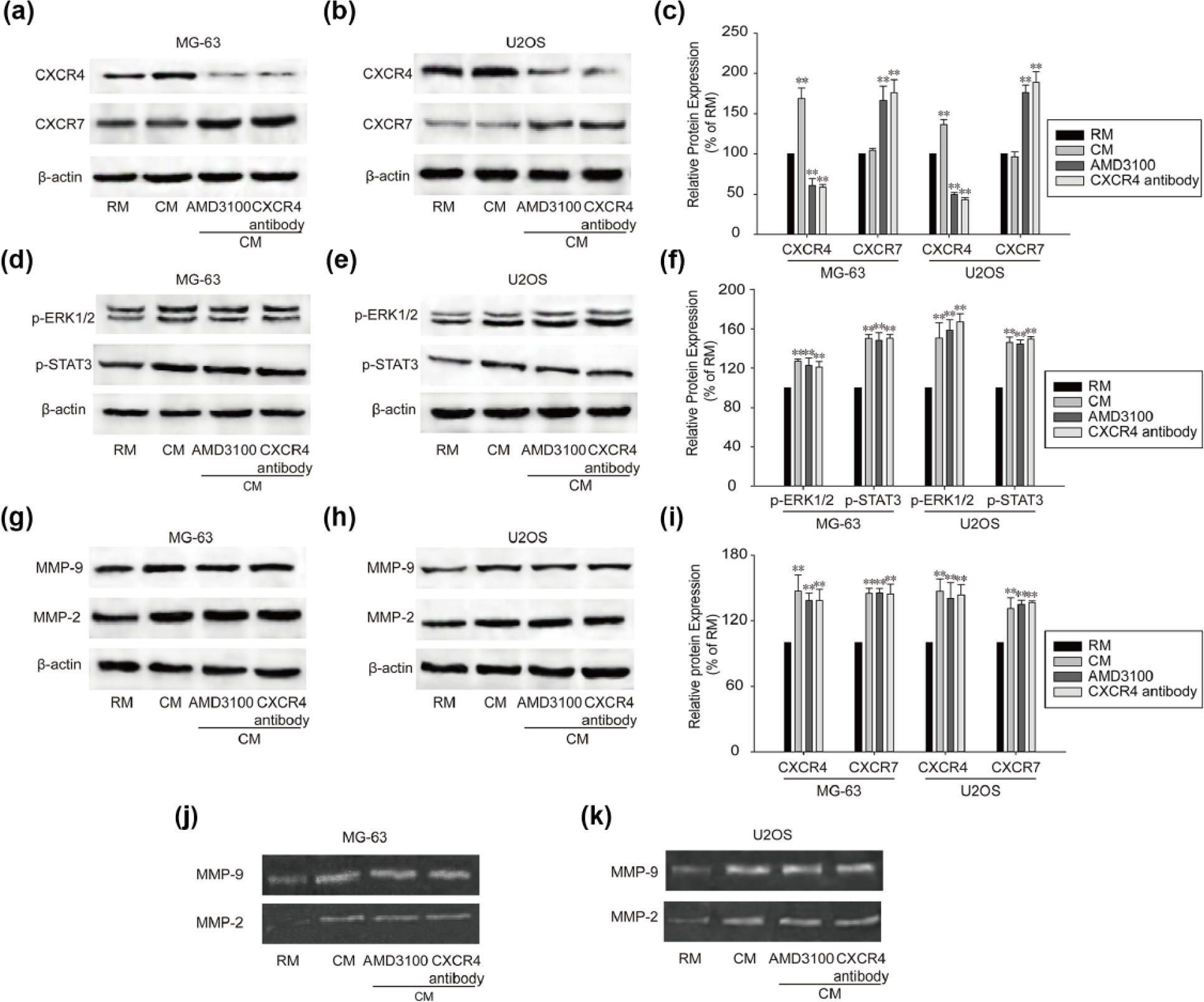
CXCR7 signal was activated after CXCR4 inhibition in OS cells. MG-63 and U2OS cells were treated with AMD3100 or CXCR4 neutralizing antibody and co-cultured with BMSCs for 72 h. (a–c) CXCR4 and CXCR7 expression in MG-63 and U2OS cells was tested with western blot assay. (d–f) Expression of p-STAT3 and p-ERK1/2 in MG-63 and U2OS cells was detected with western blot. (g–i) MMP9 and MMP2 expression was analyzed by western blot. MMP9 and MMP2 activities in (j) MG-63 and (k) U2OS cells were measured by gelatin zymography. RM stands for regular medium and CM stands for co-culture medium. Data were mean ± SD for three independent experiments (**p < 0.01 compared with regular medium groups).
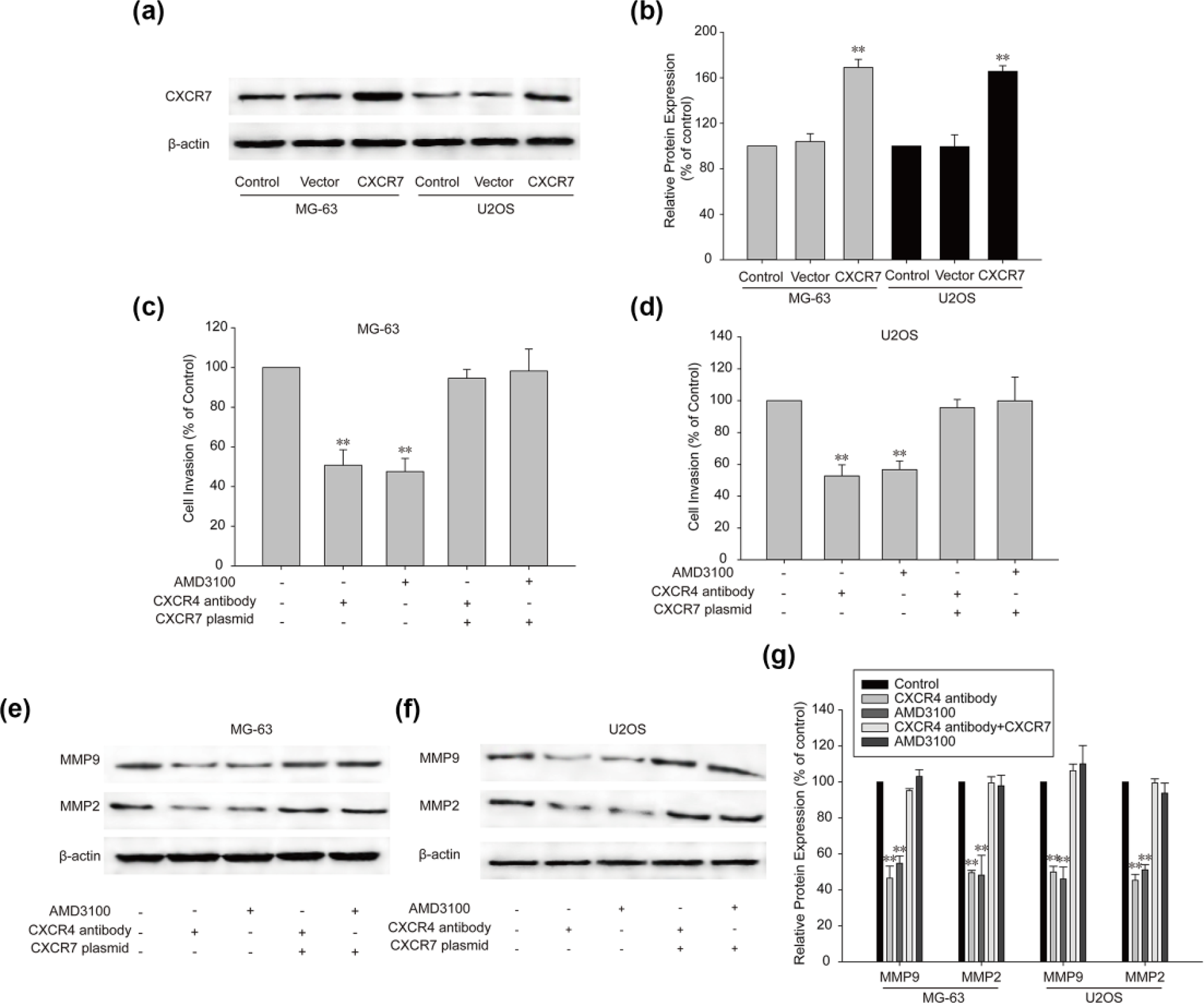
Constitutive expression of CXCR7 restored OS cell invasion which was inhibited by CXCR4 suppression. (a and b) CXCR7 expression was measured by western blot. Invasion of (c) MG-63 and (d) U2OS cells was measured with the Transwell. (e–g) MMP9 and MMP2 expression in OS cells was detected with western blot. Data were mean ± SD for three independent experiments (**p < 0.01 compared with control group).

Dual inhibition of CXCR4 and CXCR7 showed a strong effect against OS cell invasion. MG-63 and U2OS cells stably expressing CXCR7-shRNA were treated with AMD3100 or CXCR4 neutralizing antibody and co-cultured with BMSC cells for 72 h. (a and b) Protein level of CXCR7 was analyzed by western blot. Invasion of (c) MG-63 and (d) U2OS cells was determined by the Transwell. (e–g) CXCR4 and CXCR7 expression in OS cells was tested by western blot after different treatment. Expression of p-STAT3, p-ERK1/2, MMP9, and MMP2 was measured with western blot in (h and i) MG-63 and (j and k) U2OS cells. RM stands for regular medium and CM stands for co-culture medium. Data were mean ± SD for three independent experiments (**p < 0.01 compared with control group).

Bone marrow microenvironment is required for OS switch from CXCR4 to CXCR7. MG-63 and U2OS cells were treated with ADM3100 or CXCR4 neutralizing antibody and cultured in medium containing 100 ng/mL of CXCL12 for 72 h. (a–c) CXCR4 and CXCR7 protein expression was tested with western blot in both the OS cell lines. Invasion of (d) MG-63 and (e) U2OS cells was detected with the Transwell. (f–h) Cell invasion was further confirmed by testing MMP9 and MMP2 protein expression with western blot. Data were mean ± SD for three independent experiments (**p < 0.01 compared with control group; ##p < 0.01 compared with CXCL12 group).
Discussion
Interaction between BM microenvironment and cancer progression has been recognized in many reports.23,24 Understanding the way local tumor microenvironment affects tumor cells is important for choosing proper treatment strategies for cancers. This study demonstrated that BM microenvironment contributed to the activation of CXCR7 signal in OS when CXCR4 was suppressed and CXCR7 activation maintained OS cell invasion. These findings suggested that only suppressing CXCR4 might not be enough for blocking OS invasion and metastasis, and CXCR7 might play an important role in OS cell invasion and metastasis.
Recently, BM microenvironment has become a research focus in cancer research. Cancer cells affected by BM microenvironment are more likely to develop malignant characteristics such as metastasis, 25 angiogenesis, 26 and drug resistance. 27 BMSCs derived from BM tissue is abundant in the tissues adjacent to OS. 28 In this study, we found that BMSCs secreted CXCL12 and activated its receptor CXCR4 in OS cells. Activation of CXCL12/CXCR4 axis promoted OS cell invasion and metastasis.
CXCL12, also known as SDF-1α, is a secreted chemokine which is released into the interstitial space. CXCL12 acts on cells in the local microenvironment in a paracrine fashion to stimulate directional migration of hematopoietic and non-hematopoietic normal and malignant cells. 29 There are two key receptors of CXCL12: CXCR4 and CXCR7. Both the receptors were reported to be correlated with cancer progression. 30 A few studies have reported the interaction between CXCR4 and CXCR7 in cancer cells.31,32 But how they cooperate to mediate cancer development is not well understood. In this study, we used CXCR4 inhibitor AMD3100 and CXCR4 neutralizing antibody to block the function of CXCR4. AMD3100 caused CXCR4 degradation via endosomal–lysosomal proteolysis. Neutralizing antibody blocked CXCR4 function through direct binding to its receptor. Interestingly, cell invasion was not significantly reduced in BMSCs co-culture model. Furthermore, the downstream signal STAT3 and ERK was not significantly affected by AMD3100 or neutralizing antibody treatment. We hypothesized that other molecules might be activated to compensate for the loss of CXCR4 function.
CXCR7 was also reported to play an essential role in OS invasion and metastasis.33,34 Moreover, CXCR7 also binds to ligand CXCL12 and activates both STAT3 and ERK signals.35–37 We further tested expression of CXCR7 in OS cells in this co-culture model. CXCR7 was found to be upregulated when CXCR4 was inhibited. We believed that the switch from CXCR4 to CXCR7 was responsible for the sustained invasion of OS cells and BM microenvironment was the key reason of this signal switch. In regular medium, OS cells are invasive as these cells could express CXCL12 which cause the activation of CXCR4 and its downstream signals. 8 Therefore, OS cell invasion was inhibited by CXCR4 suppression in regular medium. However, no CXCR7 activation was observed which suggested the important role played by the BM microenvironment in CXCR4-to-CXCR7 signal switch.
CXCL12 showed binding affinity to both CXCR4 and CXCR7 and was targeted for cancer treatment. 38 CXCL12 was initially thought as the key factor for signal switching from CXCR4 to CXCR7 as it could activate both the receptors. We performed the experiment in CXCL12-conditioned medium. The result showed that CXCR4 expression and OS cell invasion were inhibited by AMD3100 and CXCR4 antibody treatment. However, the CXCR7 signal was not activated and invasion was not sustained in OS cells in CXCL12-conditioned medium. Therefore, CXCL12 alone was not enough for OS cell signal switch. Other factors in BM microenvironment might be responsible for the signal switch and maintenance of cell invasion. For example, CXCR7 can also bind to CXCL11 (interferon-inducible T cell alpha chemoattractant (I-TAC))12,39 or be activated by interleukin 8 (IL-8). 40 Besides the key factor that triggered such cell signal switch, there are a few more problems which need further investigations. For example, whether this signal switch also occurs in cancer cell lines other than OS cell is still unknown. Moreover, whether CXCR7 serves the same function in OS patients still needs further confirmation. Taken together, the exact mechanism explaining the interaction between BM microenvironment and OS cell invasion and metastasis still needs further investigation.
In conclusion, this research shed some light on the mechanism of invasion of OS cell in BM microenvironment. Switching from CXCR4 to CXCR7 cell signal might be responsible for maintaining OS invasion in BM microenvironment. Dual inhibition of CXCR4 and CXCR7 might be a new strategy for OS treatment.
Conclusion
In this study, we discovered that in BM microenvironment, OS cells underwent a cell signal switch from CXCR4 to CXCR7 to maintain invasion when CXCR4 was inhibited. Therefore, inhibiting both CXCR4 and CXCR7 might be a potential treatment for OS metastasis.
Footnotes
Acknowledgements
We are grateful to our lab members for their contribution to this research and their comments on the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (Grant number: 81571395).